








Statistics and Reproducibility
This study analyzes large-scale sequencing and variant annotation datasets, drawing from sources like UniRef58, UKBB17, gnomAD18, ClinVar59, and various diagnostic cohorts. Notably, no statistical methods were applied to pre-determine sample size; all available data from each cohort were incorporated unless stated otherwise in the methods section. Importantly, the experiments were not randomized, and investigators remained unblinded during experiments and outcome assessments. To evaluate reproducibility, the analysis included benchmarking across various independent datasets and comparing findings with previously published models. Public access to all code and trained models allows for reproduction of the analyses.
Data Acquisition
Multiple Sequence Alignments
Using established protocols, the EVCouplings pipeline was employed to create multiple sequence alignments (MSAs) from sequences in the UniRef100 database, sourced in March 2022.
Human Variation Data
Variants from the UKBB’s 500k release were annotated using VEP GRCh38 RefSeq and a custom annotation to optimize variant inclusion for existing models. Variants underwent quality filtering, and annotations were matched to ensure consistency between the reference and transcript sequences. In analyzing variants outside of training, genes with less than 95% coverage across UKBB participants were excluded.
Diagnostic Cohorts
All cohorts involved obtained informed consent, adhering to ethical guidelines relevant to their respective institutions. The SDD metacohort sourced de novo mutations from a combination of studies, totaling 31,058 subjects, and quality filtering was managed by the associated centers.
Clinical Variants
To evaluate predictive performance, we utilized clinically labeled variants from ClinVar, focusing on datasets from 2019 and 2020 for comprehensive analysis.
Model Building
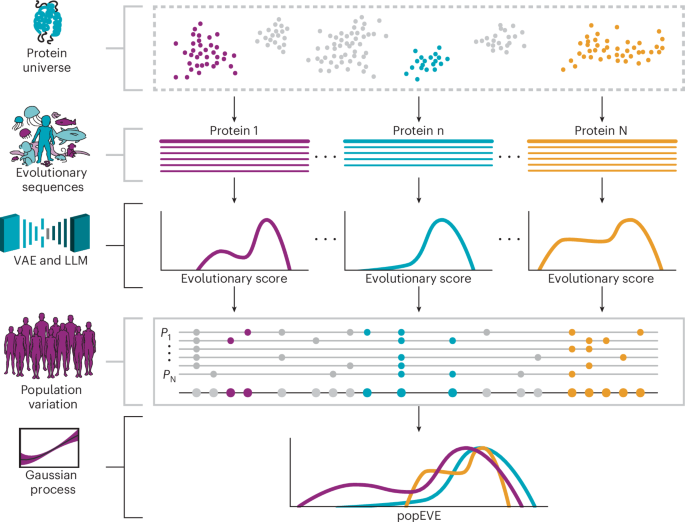
The methodological aim was to rank the severity of genetic variant effects within an individual’s proteome. The constructed probabilistic model was trained on protein sequence data from both diverse species and human populations, allowing a fusion of evolutionary and population-specific insights. This model intends to ascribe accurate impacts of variants at a single-residue resolution.
Modeling Individual Proteins
Recent advancements indicate that unsupervised models trained on protein sequences from various species successfully differentiate between benign and pathogenic variants, performing comparably to functional assays. Two model types were utilized: an alignment-based model, trained on gene-specific MSAs, and an alignment-free model derived from a full protein database.
Bayesian Variational Autoencoder
Variational autoencoders (VAEs) effectively capture complex distributions. The VAE we employed captures the conditional probability of amino acid presence at specified positions based on hidden variables. The scoring mechanism utilized the evidence lower bound (ELBO) to derive the fitness score.
Sequence Reweighting
In light of the limitations posed by phylogenetic and ascertainment biases, sequence reweighting was applied to correct for distribution irregularities. A reweighting formula adjusted sequence importance based on their alignment distance in corresponding MSAs.
Masked Language Model
The model training also incorporated ESM-1v, a language model leveraging a self-supervised masking process, enhancing its predictive capabilities for amino acid substitutions.
Estimating Variant-Level Constraint
While the individual gene models rank variants effectively, they struggle to compare variants across different genes. To tackle this, popEVE was introduced as the pioneering model for proteome-wide assessment of missense variant impacts.
Performance Assessments
Within-Protein Comparison
We assessed our model’s proficiency in ranking pathogenic variants by correlating model predictions with deep mutational scans, as well as evaluating its accuracy in predicting benign and pathogenic labels from ClinVar.
Cross-Protein Comparison
To ascertain the model’s ability to distinguish between various severity levels in clinical variants, a dataset of high-impact variants was created. This involved setting thresholds to differentiate benign from pathogenic classifications effectively.
Analysis of Patient Data
Two strategic approaches were taken to analyze de novo mutations from the metacohort, aiming to provide insights for unresolved genetic cases. Each approach, including burden testing and direct case-variant associations, was formulated to evaluate the significance of specific variants.







