








In utero electroporation
All animal procedures in this study were approved by the St. Jude Institutional Animal Care and Use Committee. In utero electroporation (IUE) followed previously established methods, where plasmids were prepared using the NucleoBond Xtra Maxi Plus EF kit from Takara Biosciences. After anesthetizing pregnant CD1 mice at E16.5 with isoflurane, the abdomen was opened to access the uterus. A cocktail of DNA plasmids, including multiple components, was injected into the lateral ventricles using a glass pipette. Electric pulses were then administered to the embryos via forceps-shaped electrodes. Specifically, six 33-V pulses of 55 ms were delivered at intervals of 100 ms. Post-procedure, the uterus was placed back, and the abdominal cavity was closed. After the pups were born, they were monitored for any signs of tumor growth, like seizures or circling, with follow-ups via MRI every two weeks. The endpoint for the study was based on defined neurological symptoms, ensuring that no limits were exceeded during the experiments. Mice designated for nucleus isolation were perfused, and tumors were preserved appropriately. Pups from at least two mothers were included for survival analysis, and randomization was not applied.
RGC isolation
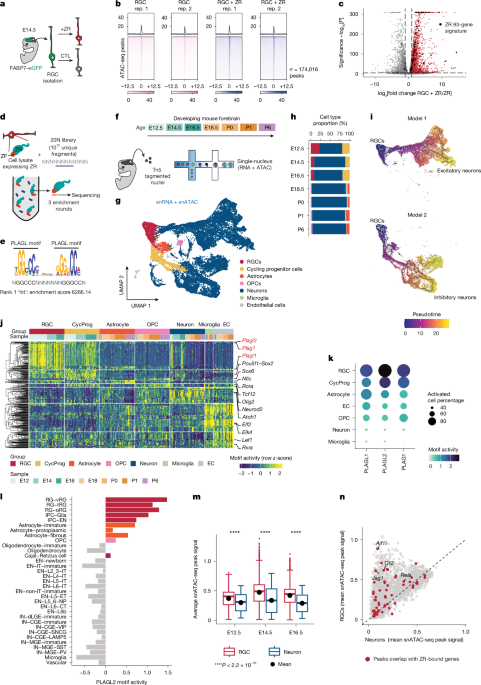
Retinal ganglion cells (RGCs) were isolated from Ink4a-knockout mice, leveraging GFP from the Blbp promoter through the Worthington Papain Dissociation system. Cells were cultivated in a neural basal medium that included various supplements and were checked for mycoplasma monthly. Culturing occurred on dishes coated with Matrigel. To create ZR-positive RGCs, they were infected with a lentivirus provided by the Viral Vector Core. Mouse tumor cells were seeded a day before this infection with polybrene to enhance viral uptake, followed by a selection phase with puromycin. ZR expression was verified through western blotting. For generating single-guide RNAs targeting Plagl1, procedures from the Center for Advanced Genome Engineering were utilized. ZR-positive RGCs underwent a similar infection and sorting process.
OPC isolation
Primary oligodendrocyte precursor cell (OPC) cultures were handled as outlined in earlier studies. Briefly, E14.5 mouse embryo cortical tissues were gathered, and neural stem cells were cultivated as neurospheres for several days before being dissociated and plated in OPC media. The cells were then exposed to either ZR-Lenti-Cherry or control virus, followed by a recovery period, after which they were collected for ATAC-seq and RNA-seq analysis.
RNA-seq and ATAC-seq analyses
RNA-seq and ATAC-seq analyses were executed using the Genialis Expression software on St. Jude’s HPC infrastructure. The RNA-seq pipeline involved steps like filtering raw reads to eliminate adaptors and subpar quality reads. Following that, STAR software was used to map reads to reference genomes, while FeatureCounts helped quantify gene expressions. Low-expression genes were filtered out before differential expression analysis using DEseq2. For ATAC-seq, the paired-end reads were processed similarly, followed by peak calling with MACS2 using specified parameters.
TrackerSeq library generation and validation
Library cloning for TrackerSeq was carried out as generally described in prior reports. This involved digesting the pCAG-SacB plasmid and inserting an 8-bit barcode using NEBuilder HiFi master mix. Post-purification, electroporation into Endura DUOs was performed, and the cells were plated for growth. Following further processing, library plasmids were purified for validation, and sequencing checks were conducted.
CSI DNA binding assay
HEK293T cells were transiently transfected with HA-tagged ZR fusion plasmids using Lipofectamine 2000. After lysing, DNA libraries containing randomized sequences were prepared and mixed for binding assays. The mixtures underwent incubation with cell lysates, and the resulting complexes were assessed through a series of washes, followed by amplification and purification of DNA library fragments.
Nuclei isolation
Tissue samples were harvested from human tumors and processed using a 10x Nuclei Isolation Kit, with some adjustments for increased lysis time for optimal nucleus quality. For mouse embryonic forebrain isolation, a different protocol was implemented. The isolated nuclei were resuspended, counted, and prepared for sequencing on a NovaSeq 6000 after library assessment.
RNAscope
Mice bearing ZR tumors underwent cervical dislocation for brain removal, followed by flash-freezing in isopentane. Sectioning was done on a cryostat, with slides stored until analysis. The RNAscope protocol followed typical guidelines, with adjustments for protease usage and fluorophore concentrations. Images were captured using a laser scanning microscope.
Immunofluorescence
For immunofluorescence, IUE surgery utilized a specific plasmid concentration. P2 pup brains were processed for fixation and embedding, followed by sectioning. Subsequent staining involved permeabilization, blocking, and incubation with primary and secondary antibodies before final mounting. Images were taken at high magnification.
snMultiome data processing
Data from human and mouse datasets were graphed using the cellranger-arc count pipeline, focusing on barcoding and quality. Moreover, reads underwent alignment to the respective reference genomes, with quality control measures in place to remove background noise. Two distinct analysis methods were utilized to filter high-quality nuclei for subsequent modeling.
snMultiome data integration
For integrating snATAC data, workflows were executed to assess all sample peaks, filtering out unsuitable peaks before quantification across datasets. The steps for determining prominence and normalization followed standardized methods to enable seamless integration across samples, leading to detailed dimensional reduction analysis.
Estimation of motif activity from snATAC-seq data
Chromatin accessibility for motif activities across various transcription factors was computed utilizing specific functions from Signac for analysis, allowing for a differentiation of motifs based on accessibility.
Cell type annotation for snMultiome and scRNA
For cell type classification, automated methods using CellTypeEstimate were employed, initially based on published marker sets, with manual revision for accuracy. Tumor samples had a suffix added to delineate tumor cell types.
Processing of TrackerSeq barcode reads
The TrackerSeq barcode reads were processed following established protocols, ensuring effective trimming and filtering of low-quality reads. A whitelist facilitated valid barcode tracking, with final barcodes matched to clean cells, categorizing them for downstream analyses.
TrackerSeq data analysis from quality control to cell type annotation
Raw TrackerSeq data was meticulously processed through a series of quality checks and filtering steps. Appropriate statistical measures ensured high-quality selections were made for comprehensive analysis, integrating the findings with other datasets for deeper insights.
Classification of malignant cells
To differentiate malignant from non-malignant cells, genome-wide copy number variations were inferred using a specific tool. Critical parameters defined losses and gains in genetic material, allowing for precise classification based on defined metrics.
ZR fusion signal signatures
The single-cell ZR fusion signature was calculated from a set of identified driver genes. A threshold score guided the classification of signal confidence, thereby enhancing the robustness of this measure in distinguishing gene expressions.
Cell cycling and non-cycling signatures
Cell cycle scoring determined cycling phases using a set methodology. Adjusting the scoring based on various markers refined the definitions of cycling versus non-cycling populations.
Construction of pseudotime trajectories
Pseudotime trajectories were modeled utilizing specific R packages, with roots based on identified progenitor cells. Cells were organized along these trajectories to allow for subsequent analyses.
Progenitor and lineage score of malignant cells
The progenitor and lineage scores were assessed by measuring the expression of shared programs. Various algorithms were used to classify differentiated cells, demonstrating the intricate relationship between gene expression and cellular identity.
Reporting summary
Additional information related to the research design can be found in the Nature Portfolio Reporting Summary associated with this article.







