








Genetic Association Analysis Summary
A recent analysis has pinpointed a recessive variant of RNU2-2 syndrome by merging data from the 100,000 Genomes Project (100KGP) with the Genomic Medicine Service (GMS) data within the National Genomic Research Library (NGRL). This analysis focused on UK pedigrees exhibiting rare disorders. Following established methods, researchers applied the BeviMed technique to compare genotypes of rare variants across various genome entries. They compared 14,805 unrelated neurodevelopmental disorder (NDD) cases against 52,861 participants without NDD. As in previous studies, only two significant genes were identified: RNU4-2 and RNU2-2.
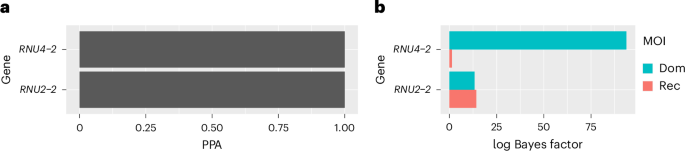
RNU2-2, notably short at 191 bp, allowed researchers to segment individuals with two rare variants, distinguishing between those in cis versus those in compound heterozygosity (i.e., trans). Rerunning the BeviMed analysis incorporating this phasing produced refined log Bayes factors, revealing minimal evidence for a recessive disorder linked to RNU4-2 but robust evidence associated with RNU2-2.
For each variant in RNU2-2, the team calculated conditional probabilities of pathogenicity under the recessive model. They identified 18 probands, termed high-confidence ‘tier 1 cases,’ with rare alleles demonstrating strong pathogenic potential. Among these, 11 were homozygous and 7 were compound heterozygous. Another 13 individuals exhibited related variants but didn’t qualify as tier 1 cases, so they were categorized as ‘tier 2.’ Additionally, none of the tier 1 or tier 2 variants were found in homozygosity in existing databases.

The analysis sought to verify the statistical connections by reviewing the assigned HPO terms of the 12 tier 1 cases. These terms showed significant similarity compared to other NDD cases. Frequently mentioned terms included ‘Generalized onset seizure’ and ‘Motor seizure,’ which were highly enriched among the tier 1 cases.
Among the tier 1 probands, most were affected by consanguinity, with several demonstrating inherited variants. Some siblings in affected families provided further evidence supporting the connections between specific genetic variants and observable phenotypes. Notably, the diverse clinical picture included conditions such as spasticity and epilepsy, often emerging in early childhood.
To replicate findings, genetic data from numerous participants across various networks was scrutinized. This validation identified several additional probands with rare variants in RNU2-2, confirming its association with recessive NDD. Specific variants from the original analysis also matched those found in this separate group.

Detailed clinical observations from several affected individuals indicated that symptoms often presented in infancy, with a spectrum of severity. Common features observed ranged from developmental delays to severe neurological impairments, with varying degrees of motor function challenges.
Interestingly, the data illustrated that RNU2-2 is the leading gene responsible for recessive NDDs identified in 100KGP, with an unusual frequency of de novo mutations suggesting heightened mutation rates. These findings point toward intricacies in the gene’s mechanics, particularly its structural and functional roles in the spliceosome.
Research further examined how deficiencies in U2 snRNA affected gene expression patterns. Lower levels of U2-2 were evident among affected individuals, coupled with significant findings correlating with U2-1 levels.

Overall, while recognizing complexities within genetic expressions, the implications of these findings have broad diagnostic potential. Understanding the variances in expression across different genes could be pivotal in differentiating between recessive and dominant disorders linked to RNU2-2 syndrome.







