








Phylogenetic analysis
The sequence of amino acids in GeCas12a2 was compared to other Cas12a2 nucleases using Clustal Omega. After alignment, it was refined with ClipKIT, allowing for the reconstruction of a phylogenetic tree via the IQ-TREE software with a maximum-likelihood method. This reconstruction involved Cas12a orthologues and used Cas12c as an outgroup. The confidence of branches was indicated by ultrafast bootstrap values, which ranged from 0 to 100.
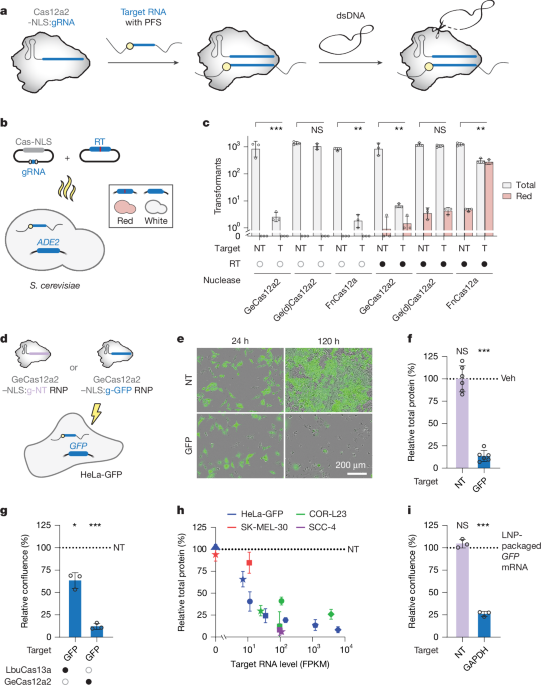
PFS library preparation
A library containing plasmids with PFS (CBS-6873) was created, featuring a target sequence followed by five random nucleotides (NNNNN). This target was positioned under a PJ23119 promoter in a low-copy sc101 origin plasmid, which houses roughly five copies per cell. The library was produced by amplifying the target-encoding plasmid using specific primers. One of the primers had a 5-nucleotide random overhang. The PCR product underwent treatment with DpnI to eliminate template DNA, was ligated, and then introduced into Escherichia coli (TOP10 strain), which resulted in over two million transformants.
PFS library depletion
The PFS preference of GeCas12a2 nuclease (CBS-6874) was evaluated through a targeted assay on the CBS-6873 PFS plasmid library, employing either a CRISPR RNA targeting the CAO1 gene or a non-targeting RNA. The sequence of GeCas12a2 was optimized for expression in E. coli. It was placed under a T7 promoter, while the crRNA was driven by the PJ23119 promoter. Escherichia coli BL21(AI) cells with both the nuclease and crRNA plasmids were subsequently electroporated with the PFS library, using about 500 ng of library DNA and 50 µl competent cells for each electroporation. The cells then recovered in lysogeny broth supplemented with IPTG and l-arabinose. Overnight cultures comprised of around two million transformants ensured significant library coverage, and plasmids were extracted via a specific midiprep kit.
PFS library sequencing and analysis
Purified plasmids from both targeting and non-targeting conditions underwent PCR amplification with specific primers. Using KAPA HIFI HotStart polymerase in a standardized manner, indexed products were sequenced on an Illumina system. The initial output was carefully processed with Trimmomatic and merged with BBMerge. Sequences with certain motifs were extracted while filtering out any with problematic bases. Depletion scores were calculated using a specific formula, and log2 fold-change values for these scores were computed across various positions of the PFS. Visual representations of PFS preferences were created using Matplotlib in Python.
Coexpression of gRNA and FLAG-tagged GeCas12a2 or FLAG-tagged FnCas12a
E. coli BW25113 cells were transformed with the GeCas12a2-FLAG expression plasmid along with a gRNA plasmid. These cells were cultivated on agar plates with antibiotics to promote selection for several hours. A colony was chosen to begin an overnight culture, which was then scaled up to 120 ml in antibiotic-rich medium. Cultivation conditions were refined, including temperature adjustments as the cells reached specific growth densities. The cells were harvested post-induction with l-arabinose.
Purification of FLAG-tagged GeCas12a2 or FnCas12a–gRNA RNP complex
Escherichia coli cell pellets were prepared and treated with a mix of inhibitors prior to being lysed. Following lysis, the supernatant was cleared and filtered. The lysate was incubated with magnetic anti-FLAG beads for overnight binding. Subsequent washes cleaned the preparation before elution employed a triple FLAG peptide. The resultant proteins were quantified and stored appropriately.
Expression of apo and catalytically dead GeCas12a2 and SuCas12a2
Both apo GeCas12a2 and SuCas12a2 were expressed using methods adapted from previous research. The gecas12a2 gene was cloned into a plasmid designed for expression, followed by transformation into appropriate bacterial strains. Cultures were grown under controlled conditions before being induced for protein expression. Cell pellets were collected for future use.
Purification of apo and catalytically dead GeCas12a2 and SuCas12a2
Cell pellets were processed similarly to prior studies for purification. They were suspended in lysis buffer and then lysed by sonication, followed by clarification through centrifugation. The lysate was bound to Ni-NTA resin for affinity purification. Subsequent washing and elution with nickel buffers were performed, with later desalting and tag cleavage for purification. Further steps included size-exclusion chromatography for clean peaks of the desired proteins, prior to storage.
FAM-labelled collateral cleavage assays
Collaterals were analyzed as previously outlined, employing specific buffer conditions. Reactions involved the proper mixing of components and were subsequently quenched for purification. Cleaved products were scrutinized through gel electrophoresis.
Yeast cell culture and quantification
Chemically competent S. cerevisiae cells were transformed by following a specialized method involving various reagents. Following transformation, cells were placed on selective agar plates and incubated, with successful knockouts being distinguished visually by color.
Mammalian cell culture
Multiple mammalian cell lines were cultured in appropriate media, supplemented accordingly and maintained to ensure viability and selection. Each line followed specific growth conditions, with careful monitoring for contamination.
Electroporation of CRISPR RNPs to mammalian cells
Cells underwent transfection with RNPs composed of various nucleases and gRNAs at set molar ratios. Specific settings tailored for different cell types were applied during electroporation. After treatment, there was an emphasis on further culture or assay preparations.
Imaging, cell counting and intensity quantification by Incucyte tracking
Post-electroporation, cells were plated for imaging and tracked via Incucyte systems over several days. Data quantification covered various metrics to assess cell behavior and response.
Total cellular protein quantification by SRB
Cells were fixed and subsequently stained before undergoing a series of washing steps to prepare for absorbance reading. The resultant absorbance provided quantification of total protein in each culture.
LbuCas13a in vitro guide verification
The activity of LbuCas13a was gauged through fluorescence measurement of substrates following incubation under defined conditions. Careful preparations were made for quantifying response to guide complex activity.
RNA-seq analysis of transcript abundance
RNA sequencing was performed on selected cell lines, with isolation and library preparation carried out according to specified protocols. Data processing was done with suitable software, which facilitated the alignment and quantification of transcript abundance.
LNP production and RNA delivery
Lipid nanoparticles were manufactured using advanced systems, and subsequent formulations were prepared for cellular application. Following careful counting of cell confluence, delivery was repeated as per experimental design.
Genomic DNA collateral cleavage assays
Assays involved treating genomic DNA under specified conditions following complex activation. Steps included genetic material preparation followed by visualization on agarose gels.
Immunofluorescence microscopy
Cells receiving GeCas12a2 or SuCas12a2 RNPs were fixed and prepared for microscopy following a detailed staining protocol. Final imaging was carried out utilizing high-quality microscopy systems.
Cell cycle analysis
Post-electroporation of RNPs, cells underwent fixation procedures for detailed analysis of genomic integrity and cell cycle progression through flow cytometry.
Annexin-V and caspase-3/7 staining
Cell analysis involved treating cultures with specific staining protocols to investigate apoptosis after RNP electroporation, followed by flow cytometry assessments of annexin V and caspase activities.
RNA-seq cell death pathway analysis
Cell deaths post-RNP electroporation prompted RNA collection for sequencing and pathway analysis. The data analysis involved high-throughput sequencing and bioinformatics tools for identifying significant differences in expression.
qPCR of incorporated dsODN for off-target double-strand-break detection
Detection of double-stranded DNA breaks was adapted from recognized methods, where samples underwent specific processing and sequencing for validation of integration sites.
Cas12a2 off-target activation analysis
To search for potential off-targets, the sequence of interest was analyzed using online tools to identify alternative binding regions, which included contextual analysis of surrounding nucleotides.
Off-target in vitro testing
A collateral dsDNA cleavage assay was performed to evaluate the specificity of Cas12a2’s response to alternative targets under controlled magnesium concentrations.
HeLa-GFP/-RFP co-culture cell depletion
RNPs were electroporated into mixed cell cultures, and subsequent analysis was performed to quantify the depletion of specific populations through flow cytometry.
Animal experiments
Slices of patient-derived cancer xenografts were transplanted into immunodeficient mice. Monitoring of tumor growth and subsequent treatment effects were carefully documented, adhering to ethical standards for animal study.
Indel enrichment
Indels were induced in specific cell lines, followed by a depletion process to refine populations for subsequent analysis of genomic alterations.
Prime edit enrichment
Cells were transfected with a combination of plasmids targeting the GAPDH locus, followed by electroporation with RNPs for precise editing assessments across the genome.
Generation of KRAS and Cas12a2-expressing stable cell lines
Viral vectors bearing coding sequences were transfected into appropriate cell lines to establish stable expression systems for further experimental work.
RNA-FISH probe creation and RNA-FISH quantification of KRAS mRNA
Custom RNA probes were designed, purified, and employed for quantifying gene expression through hybridization techniques, followed by imaging for detailed analyses.
Generation of sotorasib-resistant cells
Cells were exposed to increasing sotorasib concentrations to develop resistance, following established procedures for monitoring and analysis of surviving populations.
Statistics and reproducibility
Statistical evaluations are comprehensively presented in figure legends, with a detailed report of exact p-values and additional source data provided.
Reporting summary
Further insights into the study’s design are available to assist with understanding the research context and methodologies applied.







