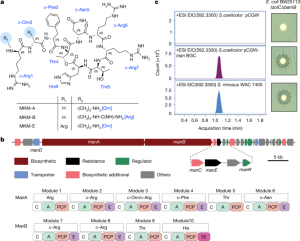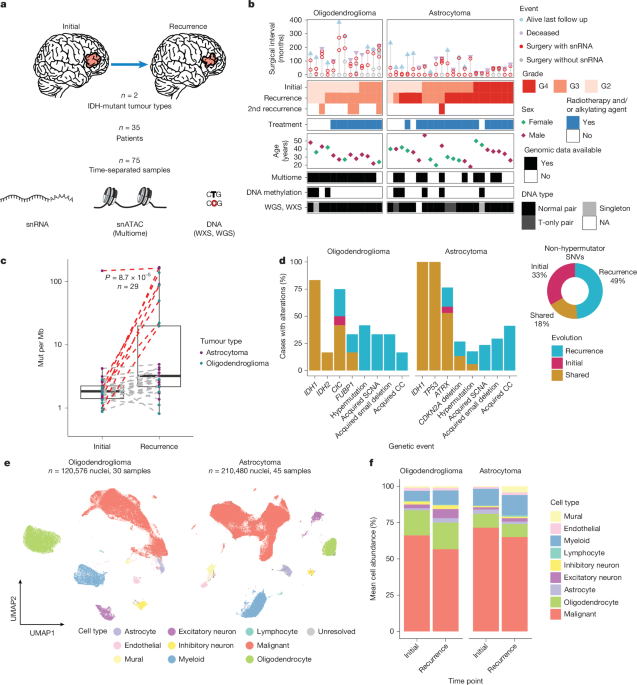
Sure, here’s the paraphrased summary of the article:
Human glioma samples were collected with consent from various institutions, including Saint Joseph’s Hospital and the MD Anderson Cancer Center. Each institution’s Institutional Review Board (IRB) approved the sample collection, with specific protocol numbers assigned to ensure compliance. Information on IDH mutation status and chromosome co-deletion was gathered from pathology reports and confirmed through DNA sequencing. Notably, astrocytoma samples showing CDKN2A homozygous deletion were noted as grade 4 according to WHO standards. Some fragmented samples were preserved for further analyses when enough material was available.
Different laboratories employed varied protocols to isolate nuclei from the frozen glioma samples, ensuring that samples from the same patient were always processed together. For instance, one laboratory used a specific buffer to dissociate samples, while another utilized an EZ lysis buffer for a gentle approach. After mechanical dissociation, nuclei suspensions were filtered and counted for downstream applications, laying out the groundwork for further genetic studies.
Bulk DNA sequencing was tailored to each tissue source, with different methodologies applied depending on the institution. For instance, the MD Anderson Cancer Center extracted DNA from glioma samples and matched blood samples using a well-known kit, while another institution used a different process for their samples. Whole-genome sequencing and exome capture were performed as needed, with several techniques ensuring thorough data collection for future analysis.
Single-nucleus RNA sequencing (snRNA-seq) involved utilizing a specific reagent kit to process nuclei, creating a unique gene expression profile for each sample. Libraries from the processed nuclei were then sequenced, allowing for comprehensive analyses of gene expression across the samples, enabling further insight into their characteristics.
The study employed various statistical methods and genomic tools to analyze genetic variants and copy number alterations from the glioma samples, ensuring that sample integrity and identity were maintained. Additional methods were used to derive functional classifications of the samples based on their genetic profiles, allowing the researchers to more clearly delineate between malignant and non-malignant states.
Through a combination of advanced methodologies, including single-cell analyses and pathway evaluations, the research aimed to elucidate the complexities of glioma samples and their unique behaviors in response to genetic mutations. Surviving analyses were conducted to understand the correlation between genetic profiles and clinical outcomes, leveraging sophisticated statistical tools throughout.
In summary, this research emphasizes a detailed exploration of human glioma specimens, with multiple methodologies employed to understand their genetic underpinnings and clinical implications in depth.