








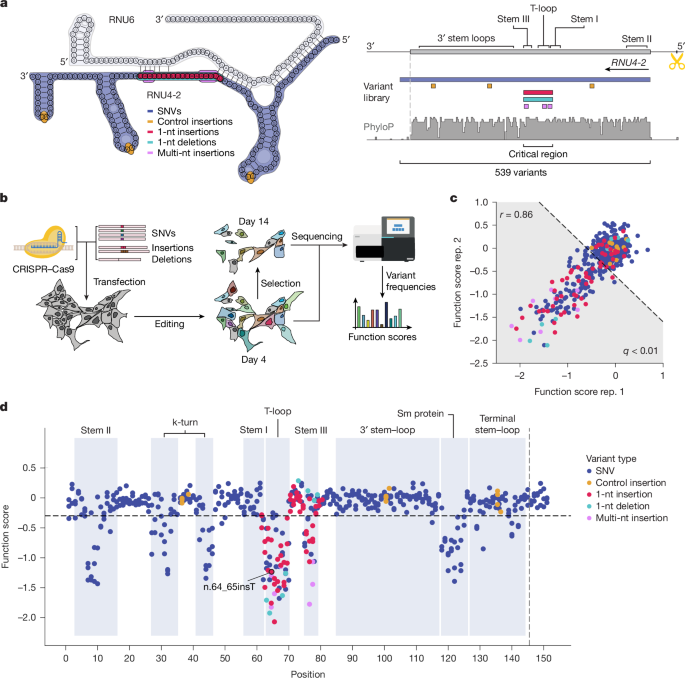
Single guide RNA design and cloning
The gRNA for SGE was designed using Benchling’s CRISPR tool, focusing on the RNU4-2 locus while considering upstream and downstream regions with low sequence similarity to RNU4-1 and its pseudogenes. A gRNA candidate was chosen based on its high on-target and low off-target scores. Notably, this gRNA was not anticipated to impact RNU4-1 because it had eight mismatches in the protospacer and PAM. The gRNA spacer sequence was then ligated into the pX459 backbone, following previous methods. Complementary primers that included the spacer were ordered, phosphorylated, hybridized, and ligated into the linearized pX459 backbone. This was followed by a PlasmidSafe DNase digestion. A 2 µl aliquot of the ligation was transformed into NEB Stable Competent Escherichia coli cells using a high-efficiency transformation protocol, and 75 µl of the transformed cells was plated on ampicillin-resistant plates, then cultured overnight at 30°C. Three colonies were selected and grown overnight at 37°C in Luria–Bertani medium with carbenicillin. The plasmid DNA was subsequently isolated using the QIAprep Spin Miniprep kit and verified through whole-plasmid sequencing. The chosen clone was cultivated in a larger volume of Luria–Bertani medium with carbenicillin, and after pelleting the cells, the plasmid DNA was extracted using the ZymoPure Maxiprep kit, purified of endotoxins, and quantified with the Qubit dsDNA BR assay kit.
HDR library cloning
An oligonucleotide library featuring RNU4-2 variants was produced by Twist Bioscience and then incorporated into a vector that had homology arms for RNU4-2, forming the HDR library for SGE.
A nested PCR used genomic DNA from HAP1 cells to create the vector with the required homology arms, targeting 700–800 base pairs flanking RNU4-2. The Kapa HiFi HotStart ReadyMix was used for PCR, and the resulting product was purified and eluted in nuclease-free water. The amplicon was then integrated into the linearized pUC19 backbone using In-Fusion HD cloning, and 2 µl of this reaction was transformed into NEB Stable cells according to specifications. The cells that formed were plated on agar containing ampicillin and incubated overnight. The resultant pUC19 plasmid with RNU4-2 homology arms was purified and sequenced from a successful clone. Following dilution, PCR amplification yielded a linearized product with compatibility to the oligo library. A PAM-blocking mutation was also introduced to minimize recutting by Cas9. The specified site for the PAM disruption aimed to reduce Cas9 activity by changing a 5′-GGG PAM to 5′-GCG. After the reaction was processed, amplification and purification occurred, leading to successful assembly of the oligo library and plasmid, which were transformed into cells using the high-efficiency protocol. Efficiency was tracked by plating a fraction of the transformed cells, with the remainder cultured for further extraction of the RNU4-2 HDR library.
HAP1 cell culture
For SGE, the use of HAP1-LIG4-KO cells—a specific line referred to as ‘HAP1’—enhanced HDR editing rates due to a mutation in LIG4. After thawing frozen HAP1 cells in a 37°C water bath, they were supplemented with pre-warmed IMDM containing various nutrients. These cells underwent a brief centrifugation, and the supernatant was removed before resuspension and plating for growth in a CO2 incubator. The media was changed the next day, establishing a routine for ongoing culture.
The subculture regimen included splitting cells to prevent excessive confluency. To do this, cells were prepared for further growth using a standard protocol involving trypsin. The washed cells were treated and resuspended in fresh medium before being recounted and replated.
Generation of diploid HAP1 cells
The parental HAP1 cells were allowed to grow for about nine days after being thawed without DAB supplementation, facilitating the natural emergence of diploid cells. After ten days, they were stained using Hoechst and then sorted to isolate diploid cells based on their G2/M peak using flow cytometry. These sorted diploid cells underwent expansion for an additional ten days without DAB prior to SGE applications.
Transfection and selection
Before transfection, cells were seeded on dishes for replicates and controls. On the transfection day, a mix comprised of the HDR library, gRNA plasmid, and polymer was prepared as per the manufacturer’s instructions. For the negative control, a different gRNA was used to inhibit successful editing, and the total volume was scaled down. Following the transfection, cells were incubated and supplied with media containing puromycin. Samples were taken at various time intervals for further analysis.
Sequencing library preparation
Genomic DNA extraction was conducted using specific kits, and concentrations were measured accurately. The RNU4-2 locus was amplified through nested PCR to prevent plasmid contamination, followed by indexing PCR to prepare samples for sequencing. Different approximations of DNA from the conditions were reacted separately, and the amplification process was closely monitored. After pooling and purifying the products, another qPCR was executed to add the necessary sample identifiers. Finally, all processed samples were combined for sequencing on a high-capacity sequencer, aiming for ample reads per experimental group.
Variant frequency quantification
The sequencing data was processed to separate and analyze variant frequencies. In essence, valid paired-end reads were filtered to exclude specific bases and duplicated entries. The HDR editing rates were assessed based on the frequency of the specific mutations identified within the sequences. Alignment against a reference sequence facilitated the determination of each variant’s occurrence.
Function score calculation
Variants maintained a frequency over a prescribed threshold, allowing for their inclusion in the detailed analysis. Function scores for each variant were calculated based on comparisons across the evaluated conditions, adjusted to account for control insertions. This analysis included statistical evaluations to address the probability of pathogenicity for each variant under specific criteria.
Variant scoring with CADD and ViennaRNA
Variants were classified based on pre-existing data, identifying pathogenic types and documenting the presence in broad genomic databases. Various analyses were conducted using CADD and ViennaRNA guidelines pertaining to structural stability for the established variants. This included assessing interaction stability between the involved RNA components while computing energetic details, further refining the understanding of how specific changes in the sequence might influence function.
Assigning evidence codes to variants based on function score
Guided by established protocols, function scores derived from SGE experiments were utilized to classify the significance of identified variants. A reference group of known pathogenic variants was established to understand the strength of the findings, alongside defining neutral variants from variant databases. Advanced statistical models were applied to predict pathogenicity probabilities for the newly identified variants.
Phenotype severity and clustering
Clinical data from affected individuals was transformed into a scale to facilitate comparisons of phenotype severity. Subsequent analyses employed PCA and UMAP methodologies to visualize relationships among clinical attributes. Statistical tests were utilized to compare variant impact based on function score classifications across the dataset.
RNA sequencing cluster analysis
RNA sequencing conducted on lymphocyte samples yielded data that enabled assessment of alternative splicing events. Following filtering and analysis protocols, significant events were retained and used for further examination of differences between affected and control groups.
Association testing in UK Biobank
Phenotypic data related to educational attainment was derived from UK Biobank, aligning specifically with cognitive scores and other relevant metrics. Statistical analyses explored relationships between variant groups and observed outcomes, accounting for age and other variables impacting results.
Investigating RNU4ATAC variants in ClinVar
A comprehensive search of ClinVar identified variants in RNU4ATAC, allowing comparisons of functional predictions and classifications based on their impact. This involved recognizing structurally equivalent sites across related genes to establish further contextual understanding.
Identifying biallelic variants in cohorts
Rare disease cohorts were scrutinized for individuals exhibiting biallelic variants in RNU4-2. The study included comprehensive prior analysis of participants’ genomic information, leading to a detailed investigation of variants known to affect neurodevelopmental disorders.
Ethics
Informed consent protocols were upheld, with ethical clearances obtained for all participant activities outlined in the study. Regulatory approvals were confirmed from multiple oversight bodies, ensuring compliance with research norms.
Reporting summary
Detailed methodological information is available in supplementary materials linked to this research.







