Cloning, protein expression and purification
The DNA for ThermoCas9, featuring a C-terminal His6 tag, was integrated into the pML-1B vector and expressed in the Escherichia coli NiCo21(DE3) strain. This involved growing the cells in Luria–Bertani (LB) medium enriched with 0.2% d-(+)-glucose at 37 °C until they reached an optical density of 0.8 at 600 nm. At this point, isopropyl-β-d-thiogalactopyranoside was added to a concentration of 0.5 mM. The cells continued growing for an additional 16 to 18 hours at 20 °C, then underwent centrifugation and were stored at −80 °C. When the need arose for previously frozen cells, they were lysed using sonication in a buffer that consisted of 500 mM NaCl, 50 mM phosphate buffer at pH 8.0 (a mix of sodium phosphate dibasic and monobasic), 5 mM imidazole, and 1 mM β-mercaptoethanol. This buffer also included 1 tablet of cOmplete Mini Protease Inhibitor Cocktail (from Sigma-Aldrich) per 100 ml. Next, the lysate was centrifuged at 16,000 rpm for an hour at 4 °C, allowing the supernatant to be transferred to a pre-equilibrated 5-ml HisTrap HP His tag protein purification column (Cytiva Life Sciences). The resin was washed with 200 ml of wash buffer containing 500 mM NaCl, 50 mM phosphate buffer pH 8.0, 30 mM imidazole, and 1 mM β-mercaptoethanol. The purified protein was then eluted using an elution buffer that included 500 mM NaCl, 50 mM phosphate buffer pH 8.0, 250 mM imidazole, and 1 mM β-mercaptoethanol. The resulting eluate was then loaded onto a pre-equilibrated HiTrap Heparin HP affinity column (Cytiva Life Sciences) and eluted using a gradient from 100 mM to 2 M NaCl. Finally, the purified protein was concentrated and stored at −80 °C for future applications.
For the purification used in human gene-editing studies, the DNA, which included a 3×-nuclear localization sequence (containing 2× SV40 NLS and 1× nucleoplasm NLS), was fused with ThermoCas9 and integrated into the pML-1B vector. This was expressed in E. coli Rosetta (DE3) cells. The purification process mirrored the earlier method, but the gel-filtration buffer was adjusted to utilize cytotoxin-free water.
In vitro RNA transcription
The T7 in vitro transcription method was used to generate the sgRNA for both ThermoCas9 and AceCas9. The sgRNA templates featuring a T7 promoter were procured from Eurofins Genomics. A 149 nt sgRNA specific to ThermoCas9 and a 106 nt sgRNA for AceCas9 were transcribed using T7 RNA polymerase in a specialized transcription buffer that included 5 mM NTPs, 50 mM Tris-HCl at pH 7.5, 15 mM MgCl2, 5 mM dithiothreitol, and 2 mM spermidine. Purification was conducted using Monarch RNA Cleanup Kits (New England Biolabs). The DNA utilized for cryo-EM and biochemical studies was also acquired from Eurofins Genomics.
Cryo-EM sample preparation, data collection and 3D reconstruction
The heparin-purified protein was mixed with sgRNA at a 1:1.5 molar ratio and incubated at 37 °C for 30 minutes, after which the resulting RNP was further purified through size-exclusion chromatography using a Superdex 200 10/300 GL column (Cytiva Life Sciences) with a gel-filtration buffer comprising 300 mM NaCl, 30 mM HEPES at pH 7.5, and 1 mM dithiothreitol. To form the Cas9–RNA–DNA ternary complex, pre-annealed substrate dsDNA was added to the RNP at a 2:1 molar ratio, supplemented with 10 mM magnesium chloride. This complex was then incubated at temperatures between 37–50 °C for 15–30 minutes. A small aliquot (4 µl) was deposited on glow-discharged Gold 300 mesh R1.2/1.3 grids, which were allowed to adsorb for 30 seconds before being blotted for 2.5 seconds under specific conditions of 20 °C and 100% humidity. These grids were rapidly frozen in liquid nitrogen-cooled ethane utilizing a Vitrobot Mark IV.
Raw micrographs capturing ThermoCas9 bound to DNA with various PAMs were collected at the Laboratory for Biomolecular Structure of the Brookhaven National Laboratory using a Titan Krios G3i cryo transmission electron microscope, while additional configurations were handled at the Pacific Northwest Center for Cryo-EM. Movies were recorded at a magnification of 105,000 in a super-resolution mode, featuring an energy filter of 15 eV, which translated to a corrected pixel size of 0.82 Å per pixel. A total dose of 50–60 e− Å−2 was distributed over 60 frames with random defocusing set between −0.8 to −2.5 µm. Motion correction was performed in bin 2 using MotionCorr2, while contrast transfer function (CTF) estimation was executed with Gctf. About 6,080 micrographs were collected, leading to the selection of 2,516,939 particles using Topaz, which were then subjected to various rounds of 2D classification via cryoSPARC, yielding 2,015,088 quality particles for 3D classification. Subsequent heterogenous refinement in cryoSPARC classified the dataset into five distinct classes, followed by multiple rounds of 3D refinement and classification using Relion 4.0 to achieve high-quality particles. This culminated in several rounds of non-uniform refinement in cryoSPARC for the final 3D structures. Structural models were constructed in COOT and refined using PHENIX to ensure satisfactory stereochemistry and real-space map correlation parameters, noting that water molecules were only incorporated based on both density and interaction chemistry in high-resolution structures.
Bacterial survival assay
The bacterial survival assay adhered to an established protocol, albeit with slight modifications. Briefly, electrocompetent E. coli BW25141 cells, accommodating the modified p11-LacY-wtx1 plasmid, which encodes the toxic ccdB protein, were transformed with 60 ng of either WT or mutant ThermoCas9 plasmids. After a recovery period of 30 minutes in LB while shaking at 37 °C, 0.05 mM isopropyl-β-d-thiogalactopyranoside was introduced, extending the recovery for another hour. Recovered cells were plated on LB agar plates supplemented with chloramphenicol (15 mg ml−1) or a combination of chloramphenicol with 10 mM arabinose. Plates were incubated at 37 °C for 16 to 20 hours, followed by manual colony counting on both plates to calculate survival rates by dividing CFUs on arabinose plates by those on chloramphenicol-only plates. To innovate ThermoCas9, a library of ThermoCas9 linker II variants was transformed into BW25141 cells containing the modified plasmid with a PAM-distal truncated protospacer of 17 nucleotides (17-mer) following the described method. CFUs from the 17-mer cells that grew on arabinose were chosen for Sanger sequencing.
In vitro DNA cleavage assay and competition assay
ThermoCas9 was mixed with sgRNA at a 1:2 ratio and incubated at 37 °C for half an hour to generate the RNP. A target plasmid at 6 nM was then incorporated into the RNP at 1 µM, allowing for varying incubation times. The reactions were halted by the addition of a 5× stop buffer made up of 25 mM Tris at pH 7.5, 250 mM EDTA at pH 8.0, 1% SDS, 0.05% w/v bromophenol blue, and 30% glycerol. Subsequent separation of reaction products was performed on a 0.8% agarose gel, followed by ethidium bromide staining.
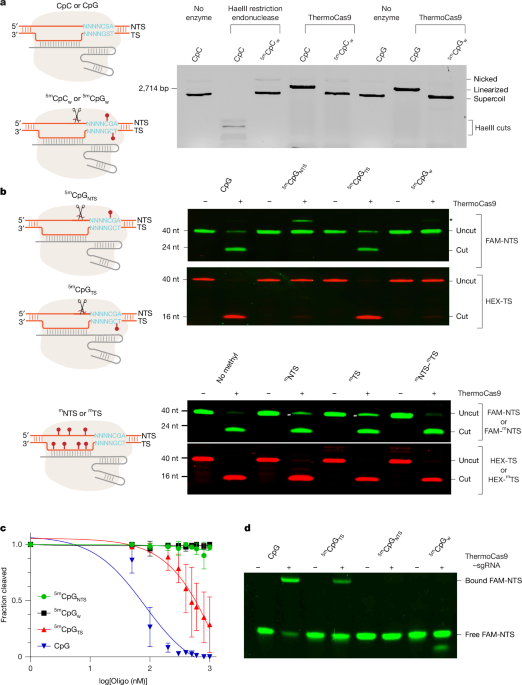
Additionally, fluorescently labelled oligonucleotides were utilized to fabricate DNA substrates. Six-carboxyfluorescein (FAM)-labelled non-target strand DNA was annealed with an unlabelled target strand DNA at a 1:1 molar ratio, while hexachlorofluorescein (HEX)-labelled target strand DNA was combined with unlabelled non-target strand DNA at a 1:19 molar ratio. Annealing involved heating the DNA mixtures to 75 °C for five minutes, gradually cooling them to room temperature. Pre-annealed dsDNA substrates were prepared to concentrations of 100–200 nM for the labelled strand and added to ThermoCas9 RNP solution at 1 µM to start the cutting reaction. Each reaction also included 10 mM of MgCl2. After incubating at 37–50 °C for one hour, 2× RNA loading buffer was introduced, featuring 97% formamide, 0.02% SDS, and 1 mM EDTA. The reaction products were then resolved through a 7 M urea 20% polyacrylamide denaturing gel while ensuring the separation of DNA fragments by size. Following electrophoresis, gel imaging was executed using a Bio-Rad ChemiDoc system, detecting fluorescent labels through excitation wavelengths of 488 nm for FAM and 580 nm for HEX.
For competition assays, ThermoCas9 RNP at 1 μM was combined with the target plasmid at 10 nM and competing oligo DNA substrates at concentrations of 50 nM to 1 μM. These reactions were incubated at 50 °C for 15 minutes and stopped with the 5× stop buffer. Products were separated on a 0.8% agarose gel and stained with ethidium bromide. The proportion of cleavage against oligo concentration was fitted to a competitive one-site binding model in GraphPad to determine the estimated binding constant of each competing oligo.
Native gel-binding assay
A mixture of FAM-labelled non-target strand DNA and unlabelled target strand DNA at a 1:1 molar ratio was prepared. dsDNA (100 nM) was blended with 1 μM ThermoCas9 RNP in the reaction buffer, excluding MgCl2, for one hour. This product was then combined with 6X purple gel loading dye and loaded onto a 10% TBE gel for electrophoresis.
In vitro methylation screening
Genomic DNA was extracted from HEK293T and HCT116 cells using the QuickDNA microprep kit. Extracted genomic DNA was incubated with 125–250 nM ThermoCas9 RNP in a reaction buffer containing 5 mM MgCl2 at 37 °C for 30 to 45 minutes. Following incubation, the reaction product was treated with E.Z.N.A. Plasmid DNA Mini Kit Solution I for ten minutes at a 1:1 volume ratio. The DNA was then cleaned using the Monarch PCR & DNA Cleanup Kit. For PCR amplification, 1 µl of the reaction product was combined with 0.25–1 µM primers and Q5 High-Fidelity 2X Master Mix. The resultant PCR product was mixed with 6X blue gel loading dye and subsequently loaded onto a 2% agarose gel alongside a 100-bp DNA ladder for electrophoresis.
In silico analysis of differentially methylated sites in human cells
Reduced representation bisulfite sequencing (RRBS) data were obtained from the ENCODE functional genomics database for various cell types. Call sets (bed files) were downloaded from the ENCODE portal with specific identifiers for the HEK293T and HCT116 cell lines. An in-house program analyzed the methylation profiles based on the gathered scores. The RRBS methylation profiles across diverse genetic loci in different cell lines were visualized using the Integrative Genomics Viewer, and differential methylation in the cell lines was examined with an in-house program based on Python scripts and bed utilities.
Transfections and gene editing in HEK293T and HCT116 cells using plasmid DNA
The human-codon-optimized thermocas9-sv40nls gene and its sgRNA module were expressed under the control of the CMV and U6 RNA polymerase III promoters. Additionally, an EGFP reporter gene was co-expressed under the elongation factor 1α promoter to facilitate sorting of successfully transfected cells. Four different spacers targeting protospacers in VEGFA, EMX1, and PRDX4 genes were designed. The differential methylation in the protospacers was noted between HEK293T and HCT116. Both cell lines maintained under specific media were seeded on treated plates at a density of 1.0 × 105 cells per well. After 24 hours, transfection was conducted using Lipofectamine 3000 reagent along with specific volumes of plasmids and media. Following 72 hours of culture, the presence of GFP was assessed through fluorescence-activated cell sorting (FACS). In an alternate approach, HEK293T cells were transfected with SpyCas9 and sgRNA in a similar manner, but genomic DNA was isolated 48 hours later without FACS sorting.
FACS
After a 72-hour incubation, transfected HEK293T and HCT116 cells underwent harvesting, followed by centrifugation and resuspension in DMEM. They were filtered through a Nylon mesh before GFP+ cells were bulk sorted using a flow cytometer. Specific gating techniques aimed to enhance green signal detection while minimizing background noise. The sorted cells were transferred to a 96-well plate and cultured for about 1 to 2 weeks until they reached approximately 75% confluency. The propagated cells were then passaged to further screening for indels.
ThermoCas9-mRNA production and nucleofection
In vitro transcription reactions for ThermoCas9-mRNA involved assembling various components including buffers, ATP, GTP, and mRNA, followed by an overnight incubation. After treatment with DNase, RNA cleanup was performed before transfections commenced. For mRNA delivery, nucleofection was carried out using a 4D Lonza nucleofector with specific conditions set for various cell types. Post-nucleofection, cells were allowed a brief resting period before being transferred to culture plates.
Screening for genome editing
Genomic DNA was isolated from non-sorted propagated cells around 2 to 3 weeks after FACS. The Zymo Research kit facilitated extraction, followed by PCR amplification of targeted genomic regions to confirm indel formation. These products were gel-purified and then sent for Sanger sequencing, with analysis done using the ICE tool. Similarly, for mRNA-treated and RNP-treated samples, genomic isolation followed the same procedures, concluding with sequencing analysis.
Bisulfite sequencing
Both HEK293T and HCT116 genomic DNA underwent bisulfite treatment as per the manufacturer’s directions. Primers to amplify bisulfite-converted samples flanking energy-targets were crafted using MethPrimer followed by sequencing.
5mC interrogation by Infinium Methylation EPIC array
DNA quantified through Qubit fluorometry underwent bisulfite conversion using a specific kit, following which it was processed per the Illumina Infinium methylation protocol. This included denaturation, amplification, and hybridization to EPIC chip beads. Scanning and subsequent analysis determined contrasting methylation scores in ThermoCas9 target sites across different cell types.
Data processing for Infinium Methylation EPIC array
The R package SeSAMe was utilized to handle microarray platform files, enabling detection of differentially methylated loci. The ‘openSesame’ function converted files into a matrix format for further analysis. Various analytical techniques pinpointed differentially methylated regions, adjusting values based on statistical significance.
Reporting summary
More detailed information about the research design can be found linked to this article.