








General methods
DNA amplification was performed using PCR, and we utilized Phusion U Green Multiplex PCR Master Mix from ThermoFisher Scientific or Q5 Hot Start High-Fidelity 2× Master Mix from New England BioLabs, except where indicated differently. Oligonucleotides came from Integrated DNA Technologies. We constructed plasmids expressing epegRNAs through Gibson assembly with a custom acceptor plasmid. The sequences of the sgRNA and epegRNA constructs employed here are detailed in Supplementary Table 1. Vectors for mammalian cell experiments were purified using Plasmid Plus Midiprep kits from Qiagen or PureYield plasmid miniprep kits from Promega, both of which include steps for endotoxin removal. All live animal experiments received approval from the Broad Institute’s Institutional and Animal Care and Use Committees. The wild-type C57BL/6 mice were sourced from Charles River (027).
General mammalian cell culture conditions
Cell lines with homozygous PTCs in TPP1, HEXA, and NPC1 were created through prime editing. We purchased HEK293T (ATCC CRL-3216), Neuro-2a (ATCC CCL-131), and HeLa (CCL-2) cells from ATCC, culturing them in Dulbecco’s Modified Eagle’s Medium (DMEM) enriched with GlutaMAX and 10% fetal bovine serum from Gibco. All cell types were kept at 37 °C with 5% CO2. Each cell line was authenticated by its supplier and tested negative for mycoplasma.
Generation of cell lines
For HEK293T cells, we seeded 100,000 cells per well in 24-well plates. After 16–24 hours, cells were transfected at around 60% confluency with 600 ng of PEmax plasmid, 200 ng of epegRNA plasmid, and 60 ng of ngRNA plasmid, following the manufacturer’s instructions for Lipofectamine 3000. Four days post-transfection, we isolated single-cell clones using limiting dilution cloning over two weeks. The selected colonies were expanded further and genotyped via high-throughput sequencing of the targeted locus, and only clones identified as homozygous for the expected edit were kept for subsequent experiments.
Lentiviral production
We transfected HEK293Ts with pMD2.G and delta8.2 packaging plasmids along with the correct lentiviral backbone using Lipofectamine 2000. The sequences for the lentiviral backbones are detailed in Supplementary Table 1. After transfection, the medium was adjusted 24 hours later. Virus-containing supernatants were collected and filtered through a 0.45-μM filter 48 hours post-transfection, and the virus was either used immediately or stored at 4 °C for up to a week.
General high-throughput lentiviral screening protocol
All oligonucleotides for high-throughput screening were sourced from Twist Biosciences as single-stranded pools. These were amplified with Q5 Hot Start High-Fidelity 2X Master Mix, creating double-stranded inserts for isothermal assembly. Specific primers used are noted in each screening section of the Methods. Typically, 11 to 13 PCR cycles were utilized for amplification, and the size of the double-stranded inserts was checked using TapeStation. For each 20-μl reaction designed for cloning into various lentiviral backbones with a 300-bp insert, we combined 50 ng of the respective lentiviral backbone with 10 ng of the relevant double-stranded insert using NEBuilder HiFi DNA Assembly Master Mix. The number of reactions was scaled according to the library’s size, with additional reactions set up for every 1,000 elements. Reactions were incubated at 50 °C for two hours, pooled, and purified via the QIAquick PCR purification kit, with yields typically eluted in a minimum of 20 μl of ddH2O or 2.5 μl per original reaction.
The isothermal assembly reactions were electroporated into NEB 10-beta Electrocompetent Escherichia coli and plated on LB plates to achieve at least 1,000× coverage of each library element. After 14 hours, colonies were scraped and prepared using Qiagen Plasmid Plus kits, and pooled plasmids were subsequently transfected into HEK293Ts alongside lentiviral packaging plasmids. The medium was changed 24 hours post-transfection, and virus-containing supernatants were collected and filtered through a 0.45-μM filter 48 hours later. The cells were then transduced with virus at a multiplicity of infection of 0.3, aiming for at least 1,000× coverage. Two days later, they were passaged with puromycin to enrich transduced cells. For experiments including FACS isolation of GFP-positive cells, we determined coverage by multiplying the percentage of GFP-positive cells with the number of library elements, often sorting at least 50,000 cells for a 500 element library. We pelleted and froze cells on dry ice, and genomic DNA was extracted using QIAamp DNA Micro or Mini kits, depending on the number of isolated cells.
In the initial PCR reactions, all genomic DNA for each biological replicate was utilized, employing Q5 Hot Start High-Fidelity 2X Master Mix to amplify the integrated lentiviral cassette and append sequencing adapters. For each reaction, we processed a maximum of 5 μg of genomic DNA. Primers are indicated within the screening sections and in Supplementary Table 1. Subsequent PCR reactions were purified, and a portion was utilized for a second round of amplification, adding unique sample indices and flow cell adapters to each amplicon. Final reactions underwent bead purification and quality check via TapeStation, with quantification carried out through Qubit before high-throughput sequencing on an Illumina MiSeq, NextSeq, or Element Biosciences AVITI instrument. Specific sequencing conditions and analyses were documented for each screen.
PE2 screening
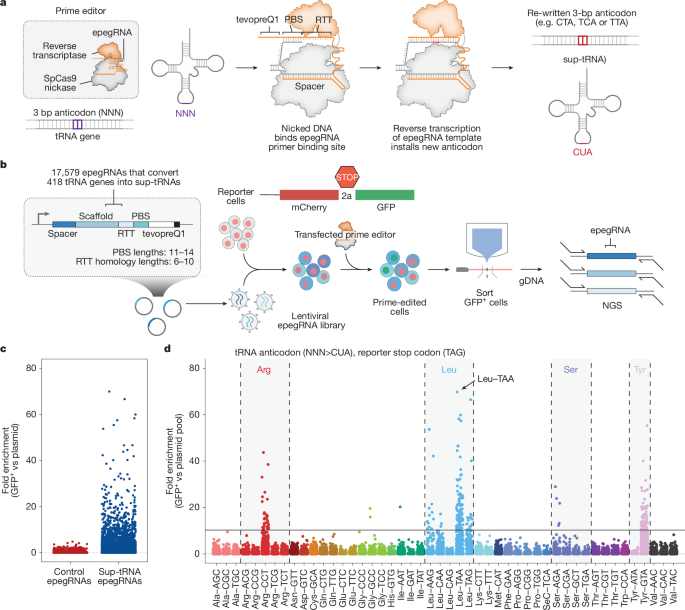
To convert endogenous tRNAs into sup-tRNAs, we first pinpointed 418 high-confidence tRNA genes and selected two nearby 20-bp spacers featuring an NGG protospacer-adjacent motif surrounding the anticodon. When necessary, we allowed prime editing to target multiple tRNAs with identical mature sequences. We modified PBS lengths from 11 to 14 bp and adjusted RTT homology lengths in the 3′ region beyond the last edited nucleotide from 6 to 10 bp. Three distinct prime editing screens were created, with RTTs replacing the anticodons of endogenous tRNAs with one of three sup-tRNA anticodons (CUA, UCA, or UUA). To boost prime editing efficiency, a structured RNA motif called tevopreQ1 was added at the 3′ end of each pegRNA, resulting in the formation of epegRNAs. Overall, we designed 17,579 epegRNAs aimed at converting endogenous tRNAs to sup-tRNAs for all three aforementioned anticodons. This pool also featured 420 control epegRNAs targeting serine and arginine tRNAs, switching their anticodons for different serine and arginine anticodons that are not anticipated to yield a signal in a readthrough-based screen. A unique barcode sequence was attached after the polyT terminator for each epegRNA to help assign each sequencing read to the proper element. The finalized oligonucleotides were constructed with a common 5′ end for isothermal assembly, a unique epegRNA sequence, a polyT for Pol III termination, an adjustable-length linker, a unique 25-bp barcode, and a common 3′ end for isothermal assembly. Prior to isothermal assembly, oligonucleotides were amplified, followed by sequencing on an Illumina MiSeq with specific primers utilized for read alignment. Analysis compared normalized reads based on sequencing depth, utilizing library representation as a foundational comparison.
Lentiviral sup-tRNA screen
We assessed a range of tRNA promoter variants by swapping anticodons in 418 high-confidence human tRNA sequences to CUA, UCA, or UUA to target stop codons TAG, TGA, or TAA, respectively. Each tRNA variant was assigned a unique barcode to mitigate sequencing error impacts on library member assignments. We also included a pre-integrated Nextera Read1 adapter for seamless reading of cluster-defining barcodes. We cloned this variant pool into three lentiviral backbones to investigate the influence of distinct upstream elements on sup-tRNA effectiveness. The first 25 read cycles were precisely matched with the respective barcode per pool element for analysis. Normalization was performed according to sequencing depth, comparing sorted samples against the plasmid pool representation for each element.
Leader and terminator sequence screening
Initially, we screened for leader sequences using the 40 bp upstream of each endogenous high-confidence tRNA in the human genome, placing these upstream of a tRNA-Leu-TAA-3-1 sequence modified to include CUA as its anticodon, following with a polyT terminator. Control sequences shared the same leader sequences but kept the native anticodon. Sequencing data utilized exact matches for both leader sequences and anticodon regions for correct library assignment. Additional details are logged in Supplementary Table 4.
To widen the scope of leader sequences incorporating multiple sup-tRNAs, we fashioned a lentiviral library containing six leaders (two optimal, two suboptimal, and two random) with 418 human tRNAs, their anticodons swapped for CUA, UCA, or UAA. We assessed the effects of terminator sequences placed downstream of sup-tRNAs with their corresponding leader sequences, resulting in a total of 11,543 combinations. Each tRNA included a unique 20-bp barcode for ease in sequencing read assignments. Reads were matched accurately for respective elements in the pool.
Saturation mutagenesis screening
We engineered variants for saturation mutagenesis screening by generating every sequence containing a SNV, paired substitutions at all hairpin positions, and 1-bp deletions computationally. To facilitate assembly, 25-bp ends were attached to either side. Each tRNA variant was assigned a unique barcode to minimize sequencing error preferences affecting library member assignments. A pre-integrated Nextera Read1 adapter was also included for accurate sequencing alignment. Variants specifically for tRNA-Leu-TAA-4-1, tRNA-Arg-CCT-4-1, mouse tRNA-Leu-TAA-2-1, and tRNA-Tyr-GTA-2-1 were ordered as Twist oligonucleotide pools. A comprehensive pooled library was also ordered targeting tRNA-Leu-TAA-1-1, tRNA-Leu-TAA-2-1, tRNA-Leu-TAA-3-1, and tRNA-Leu-TAA-4-1.
For experiments involving tRNA-Arg-CCT-4-1, mouse tRNA-Leu-TAA-2-1, and tRNA-Tyr-GTA-2-1, we incorporated leader sequence elements for each tRNA within the Twist oligonucleotides, which were then cloned into the pSEP0310 lentiviral backbone. In contrast, for the tRNA-Leu-TAA-4-1 and Leu-TAA focused library, oligonucleotides lacked leader sequences and were incorporated into the pSEP0308 backbone with an exogenous hU6 promoter. Libraries underwent sequencing on an Illumina instrument covering respective barcode and tRNA regions. For analysis, read alignments confirmed barcodes against tRNA elements.
epegRNA optimization with synthetic target-site screening
To refine epegRNA structures, we developed a lentiviral library of 17,280 epegRNAs, exploring five spacer variants, PBS lengths from 8 to 16 nt, RTT lengths between 21 to 36 nt, and various combinations of the targeted mutation variants alongside 720 control epegRNAs that did not alter anticodon edits. Each oligonucleotide encoded a synthetic target site adjacent to the lentiviral epegRNA, allowing us to assess outcomes of both the epegRNA and editing in the same sequencing read. Following transfections with a panel of prime editor proteins—including PEmax and engineered PE6 variants—we performed experiments in both MMR-deficient and MMR-proficient cells, along with scenarios where MMR inhibition occurred via co-transfection of dominant-negative MMR proteins. For HeLa cells, genomic DNA was collected three days post-transfection, while for HEK293T cells, it was gathered at both three and nine days. We carried out high-throughput sequencing of the lentivirally integrated cassette, aligning sequencing reads with the corresponding epegRNA library.
RNA-seq and transcriptomic analysis
HEK293T cells were transfected using our optimized epegRNA and ngRNA alongside PE6c or with a contrasting epegRNA and ngRNA targeting the HEK3 locus with PE6c. We extracted RNA six days after transfection through the Qiagen RNeasy kit. Following extraction, we constructed RNA-seq libraries using the SMART-Seq mRNA LP kit and sequenced them with 2× 75-bp on an Element Biosciences AVITI instrument. Adapter sequences were trimmed from the fastq reads, aligned to the human genome with STAR, and differential expression analysis was executed using DESeq2 and custom scripts.
Measurement of tRNA abundance
Total RNA, including small RNAs, was isolated from prime-edited cells using miRNeasy kits. RNA was reverse transcribed with SuperScript IV, using primers tailored to each tRNA gene family under examination. Thereafter, quantitative PCR was conducted with Power SYBR Green PCR master mix and primers specific to each family. For tRNA-Leu-TAA-1-1, targeted tRNA sequencing was performed where we quantified the desired tRNA edit relative to unedited versions across a polyclonal edited population, in both genomic DNA and total RNA.
Sequence context screening
To create a diverse PTC sequence context reporter library, we identified 14,746 naturally occurring premature TAG stop codons in protein-coding genes flagged as pathogenic, likely pathogenic, or uncertain in ClinVar. Each TAG codon was flanked by 18 nucleotides of its native mRNA sequence. Positive controls maintained the same context but swapped the TAG for a TTG Leucine codon, while negative control members, totaling 2,800, were drawn from ClinVar variants along with TTG controls, with modifications to ensure readthrough prevention.
Using oSEP0163 and oSEP0164, we initially linearized the pSEP0211 lentiviral backbone, amplifying the oligonucleotide pool via oSEP0165 and oSEP0166. These sequences were then integrated into the pSEP0211 backbone between mCherry and GFP through Gibson assembly. Two days post-transduction, we extracted mRNA and genomic DNA from the cell population, underwent reverse transcription, and sequenced the reporter construct from both cDNA and genomic samples.
For analysis, we derived an ‘RNA score’ metric by calculating each element’s frequency in cDNA compared to that in genomic DNA. To account for variations in transcript expression levels due to unique contexts, we formulated a ‘readthrough score’ for each ClinVar PTC by comparing RNA scores with their no-premature-stop equivalents.
Off-target prime editing screening
To discover potential off-target sites for prime editing, we used Cas-OFFinder to identify all human genomic sequences that could have up to six mismatches or a combination of four mismatches with two bulges relative to our optimal epegRNA spacer sequence. We included additional local sequences around targeted sites for context. Our positive controls represented unique barcoded sequences corresponding to the endogenous tRNA-Leu-TAA-1-1 site, while negative controls without spacer sequence homology were fabricated and transduced into HEK293T cells. These cells underwent transfection with either an epegRNA alone or with a PE6c prime editor expression plasmid. Genomic DNA was then isolated three days later, augmenting the lentiviral integrated target site for sequencing.
rhAmpSeq off-target-site amplification and analysis
Initially, we used Cas-OFFinder to identify all human genomic sequences with a max of 4 mismatches for our optimum epegRNA spacer. Pooled sequencing primers for selected human off-target sites were created employing the rhAmpSeq design tool. Following DNA extraction from editor-treated HEK293T cells, pooling sequencing primers were used for amplification. Amplified libraries underwent sequencing with 300-bp single-end reads on an Illumina MiSeq, where sequences were extracted using the R Bioconductor BSGenome package, aligning them to amplicon reference sequences, with evaluation of read quantities representing the potential edits.
Flow cytometry
Cells were trypsinized, re-suspended in a medium with 10% FBS, and filtered through a 45-μm cell strainer before performing flow cytometry or FACS isolation. Flow cytometric analysis utilized the CytoFLEX LX Flow Cytometer and CytExpert Software at the Broad Institute Flow Cytometry Core. FACS isolation was executed on the SONY MA900 Cell Sorter, after which the cells were sorted into medium and then centrifuged.
General cloning
Cloning of plasmid vectors for epegRNAs or ngRNAs in mammalian expression was achieved through isothermal assembly. This process involved linearizing a human U6 promoter vector via PCR and incubating it with IDT eBlocks that encode full-length epegRNA or ngRNA sequences necessary for isothermal assembly using NEBuilder HiFi DNA Assembly Master Mix. A list of used epegRNA and ngRNA sequences can be reviewed in Supplementary Table 1.
AAV vector genomes were assembled following the same isothermal assembly techniques outlined previously. Briefly, the v3em vector genome construct was linearized, and new epegRNA and ngRNA sequences were inserted via isothermal assembly using NEBuilder HiFi DNA Assembly Master Mix. The final v3em vectors were assembled through similar methods.
Arrayed genome-editing experiments
Experiments using HEK293T or HeLa readthrough reporter polyclonal cell lines, along with Neuro-2a cell testing, involved seeding 12,000 cells per well into 96-well plates and transfecting the following day with Lipofectamine 3000. Each well received distinct amounts of prime editor plasmid, epegRNA plasmid, and ngRNA plasmid. Three days post-transfection, cells were lysed, and genomic DNA was gathered using a lysis buffer.
For the HEK293T disease model rescue experiments, cells were first seeded at 200,000 per well in 12-well plates and transfected the next day with a specific amount of each plasmid. Genomic DNA was then extracted after three days of incubation similar to prior procedures.
Protein isolation and enzymatic activity assays
Human cells were collected by trypsinization, pelleted, and washed. For the TPP1 assay, cells were lysed in a Triton X-100 and SDS buffer at 4°C. The HEXA assay used RIPA homogenizing buffer for lysing at the same temperature. The resulting lysates underwent centrifugation, and the supernatant was used for total protein quantification.
For the TPP1 assay, a specific amount of protein lysate was incubated overnight in a designated substrate buffer, with fluorescence measurement being taken afterward to assess enzymatic activity relative to wild-type cells.
In the case of the HEXA assay, another protein lysate volume was used in a similar fashion, with reactions halted and fluorescence measured to derive activity coefficients relative to wild-type cells.
For analyzing α-L-iduronidase in mouse tissues, we homogenized samples, then extracted proteins for concentration quantification, followed by incubation in a substrate buffer to measure endpoint fluorescence.
Western blotting
Post-collection via trypsinization, cells were subjected to washing, lysis, and centrifugation to extract supernatants for total protein quantification. We separated protein lysates via SDS–PAGE and transferred them onto nitrocellulose membranes. Following membrane blocking, primary antibodies specific to NPC1 or GAPDH were applied, followed by secondary antibodies for signal detection. Ultimately, we quantified signals according to standardized protocols.
Protein isolation, trypsin digestion and TMT labelling for mass spectrometry
Protein isolation was previously outlined for Western blotting. Samples were processed through S-trap micro spin columns with specific alterations for trypsin digestion under careful conditions. The resultant peptides were prepared for tandem mass tag (TMT) labelling as per the manufacturer’s directives, followed by reaction quenching and combining labelled samples. These samples were then speed-vacuumed.
LC–MS
We utilized reverse-phase HPLC for TMT-labelled tryptic peptides with specified solvents under a defined gradient before conducting nano-electrospray mass spectrometry. The full MS and MS/MS cycles were executed across set parameters.
Database search with Proteome Discoverer
We utilized raw mass spectral data files for searching using specified parameters in Proteome Discoverer through Sequest HT, accounting for several modifications.
AAV production
AAV production employed HEK293T clone 17 cells in specific culture conditions. Following a one-day incubation and transfection with designated plasmids, cells were collected for a series of precipitation and lysis steps to isolate viral components. The collected solutions went through ultracentrifugation, followed by a careful extraction and buffer exchange, concluding with sterile filtration and quantification.
Animal use
All animal experiments received prior approval from the relevant institutional committees. Mouse housing adhered to stipulated environmental standards with comfortable living conditions provided for the study.
Neonatal ventricular injections
Microinjection syringes were prepared and filled with a specific injection solution. Mice were anesthetized before undergoing injections in monitoring conditions to ensure precise delivery. Gender balance was maintained throughout all experimental conditions without pre-specified statistical calculations for group sizes.
In vivo prime editing
We evaluated stop codon readthrough through AAV delivery involving designed epegRNA structures for both TAG and TGA codon conditions. We examined long-term impacts of PERT by introducing required editing reagents and assessing the results through mass spectrometry analysis of extracted tissue.
Mouse tissue collection, histology and immunohistochemistry
Mice were sacrificed, and tissues were promptly dissected and frozen. We extracted the necessary proteins, genomic DNA, and RNA using a well-defined protocol. For histological analysis, tissues were embedded and sectioned, staining followed specific techniques for distinct cellular components. Histopathology evaluations were conducted by qualified professionals.
Statistics and reproducibility
All screens underwent independent biological replicates, sample sizes being documented and comparisons indicated. Relevant statistical values can be found in supplementary materials.
Reporting summary
For further information on the research design, please refer to the Nature Portfolio Reporting Summary linked to this article.







