








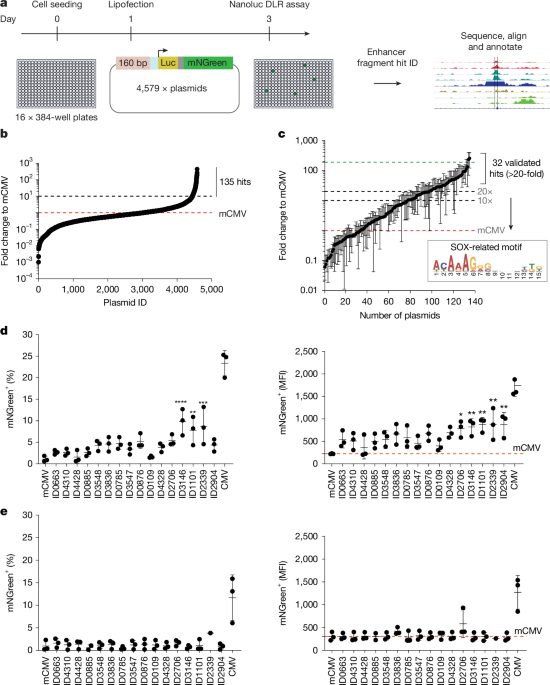
Destination vector cloning
We developed a custom destination vector specifically for Golden Gate cloning of enhancer fragments. This vector included the reporter gene NanoLuc-Ires-mNGreen-pA, positioned downstream of a mCMV promoter. To facilitate the assembly and selection process, a bacterial suicide ccdB cassette was placed at the enhancer position, which allowed for efficient cloning. This was based on the established EMMA Golden Gate cloning system. For the long-term differentiation experiments using GSCs, the same vector was modified to incorporate PiggyBac transposase recognition sites around the whole cassette.
Bioinformatics to design a SOX2 enhancer oligonucleotide pool
We revisited some published SOX2 ChIP–seq and H3K27ac data from GSC cell lines. The goal was to pinpoint GSC-specific SOX2 peaks that coincide with H3K27ac while being absent in differentiated cells (those grown in serum conditions). After identifying these peaks, we combined them and manually annotated the results to exclude centromeres. This process yielded 1,721 peaks averaging 402 bp in length, which we then used to create a set of 160 bp sequences. We added 20 bp adapters to the edges of these sequences, synthesizing them into an oligonucleotide pool via Twist Bio.
Construction of an arrayed plasmid library of enhancer fragments
The oligonucleotide pool, containing 9,523 unique sequences, was amplified through 10 cycles with a specified volume, and a portion of this reaction was utilized for an additional 15 amplification cycles to prevent PCR ‘jackpot’ amplification. The final products were cloned into an expression vector through a Golden Gate reaction. We employed KAPA HiFi Hotstart polymerase with GC buffer and adjusted temperatures appropriately. Ultimately, we picked 4,579 individual plasmids, prepared them, and organized them into an arrayed plasmid DNA library on 96-well plates. This number was considered optimal for library screening. To minimize loss, we employed a magnetic-bead-based SPRI purification method before the cloning process. Random Sanger sequencing from selected plasmids confirmed that the samples were diverse and traceable back to the original library design.
Screening platform for 384-well plates
Cells were seeded into 384-well plates with a multidrop method, and the transfection occurred the day after using CyBio Felix. After two days, we conducted a NanoGlo DLR assay to identify hits that exhibited a fold change to mCMV of over 10. Those hits were then Sanger sequenced to deduce their specific sequences and subsequently matched back to the genome to verify their overlap with the original SOX2 ChIP–seq data. The GREAT tool was used to predict the target gene for these hit sequences.
General cell culture procedures
The GSC lines GCGR-E17, E21, E27, E28, E31, E34, E37, and E55, alongside human NSC lines NS9FB_B, NS12ST_A, and NS17ST_A, were generated in our lab and can be requested from the Glioma Cellular Genetics Resource. We secured informed consent for using patient tissue, and all procedures involving patient brain tissue were approved by the NHS Health Research Authority and relevant ethics committees. Cells were cultured as adherent monolayers under serum-free conditions, adhering to established protocols. Mycoplasma testing showed negative results at passage 3. Authentication of the cell lines was confirmed using STR profiling services.
For lipofection, Plus reagent and Lipofectamine LTX were each mixed in a reduced-serum medium, then combined with DNA samples. After a brief incubation period, this mixture was carefully applied to the cells, which were analyzed two days post-transfection.
HEK293 cells were seeded at the desired density, transfected the following day with the appropriate mix, and analyzed two days later.
Screening assay using the Nano-Glo Dual-Luciferase reporter assay system
This assay involved two steps. Initially, a normalization plasmid PGK-Firefly-Luciferase was co-transfected with the experimental plasmid. Measuring firefly luciferase activity first allowed normalization of transfection efficiency. After washing cells with PBS, cell lysate was prepared for analysis. The subsequent NanoLuc reaction was run, and readings were obtained, allowing data normalization against the Firefly reaction. This helped calculate changes relative to a control vector.
Immunocytochemistry
Cells were subjected to PBS washes and fixed in 4% paraformaldehyde, with subsequently permeabilized cells undergoing blocking and antibody incubation. Following washes, secondary antibodies were administered. The cells were then stained with DAPI for nuclear visualization. Images were acquired with a Nikon TiE microscope.
Flow cytometry
For flow cytometry, cells were processed to obtain a single-cell suspension and stained for viability analysis. Data were acquired using a BD LSRFortessa cell analyzer and analyzed using appropriate software.
RNA isolation, cDNA synthesis and RT–qPCR
RNA was extracted using a commercial purification kit, then the concentration was measured. cDNA synthesis followed strict protocols, utilizing set amounts of RNA. RT–qPCR was executed using well-known methods and underwent careful data analysis to ensure reliability, focusing particularly on normalization against housekeeping genes.
Western immunoblotting
Protein lysates were prepared and quantified, followed by SDS-PAGE and transfer onto membranes for western blotting. Specific antibodies were used for visualization, and the resulting blots were analyzed for expression levels.
Precipitation of interacting proteins
Using PCR amplification and purification processes, templates were generated that facilitated protein pull-downs. Enhancer fragments were amplified with biotinylated primers and subsequently purified.
SOX2 and SOX9 ChIP–seq library preparation and analysis
After treating GSCs with formaldehyde for crosslinking, chromatin was fragmented and antibodies were used for immunoprecipitation. Following careful wash and elution procedures to isolate the chromatin, sequencing libraries were prepared and analyzed, ensuring diverse representations of the primary GSC lines.
scRNA-seq sample preparation
On the day cells were seeded, they were properly counted and arranged in plates for optimal growth. Subsequently, AAV vector stocks were introduced to the culture medium to ensure high transduction efficiency. Cell fixation and processing for single-cell transcriptome sequencing were carried out meticulously.
scRNA-seq library preparation
Libraries involving numerous samples were generated, following specific barcoding and amplification processes essential for sequencing. Quality control steps were diligently implemented before moving on to analysis.
scRNA-seq analysis
The sequencing reads were aligned, and various filtering steps were undertaken to generate reliable datasets for further analysis. This analysis also included the introduction of additional genes to capture specific cell responses during the experiment.
SCENIC analysis of scRNA-seq data
Post-filtering, the dataset was organized for advanced analysis focusing on transcription factor modules and their enrichment across different libraries. This work highlighted key factors relevant to the study.
Lentivirus production and titration
In producing lentiviral samples, HEK293T cells were cultured and transfected, followed by viral collection and purification steps to ensure effective supernatant generation.
Overexpression of SOX2 and SOX9
Fibroblasts underwent specific transduction processes to enhance the expression of SOX proteins. Following transduction, subsequent induction steps were enforced to achieve overexpression outcomes.
Nuclear extraction of GSC7 and hFib cells
Both GSC7 cells and fibroblasts were prepared for nuclear extraction procedures, with careful homogenization and centrifugation steps to isolate nuclei for future analyses.
Western blot analysis of overexpression
The protein concentrations from lysates were assessed, and overexpression verification involved rigorous western blotting techniques to confirm protein presence and levels.
Recombinant SOX protein production
His-tagged SOX2 and SOX9 were expressed and purified from bacterial cultures, using specific buffers and methods tailored for protein isolation and subsequent analysis.
Small-molecule inhibitor screening
Inhibitors were screened using various concentrations, with cells analyzed after treatment to determine their efficacy and resulting impacts on cellular processes.
EMSAs
Electrophoretic mobility shift assays were conducted to analyze binding of various nuclear extracts to specific DNA sequences, using Cy5-labeled probes in well-defined experimental setups.
Adult human cortex and brain tumour slice cultures
Brain tissues were carefully prepared for slice cultures, ensuring optimal conditions for cell viability and subsequent experimental procedures.
Addition of virus
Viruses were introduced to the tissue slices at regular intervals, followed by incubation to allow for adequate internalization and subsequent analysis.
Immunostaining
Post-incubation, slices underwent immunostaining for analysis, including blocking, primary, and secondary antibody applications, followed by thorough washes and imaging.
Zebrafish experiments
Embryos were prepared for genetic experiments, focusing on neural progenitor cell interactions after specific injections and treatments to understand developmental effects.
AAV medium-exchange transduction assay
Cells underwent transduction processes, aiming for optimal conditions to facilitate AAV incorporation, followed by analysis to measure effectiveness.
Prodrug treatment
Prodrug treatments were administered with careful preparations and controls to monitor impacts on cellular outcomes.
Incucyte live-cell imaging
Live-cell imaging was utilized to observe changes and cellular dynamics throughout treatment protocols, facilitating real-time analysis.
MTT assay
The MTT assay process involved adding the dye to cells, followed by incubation, and subsequently measuring absorbance to evaluate cell viability.
Statistical analysis
Data analysis incorporated multiple software platforms to ensure accuracy and reliability, documenting findings with appropriate visual representations.
Tumour initiation by transplantation and intratumoral AAV delivery
All animal research adhered to ethical standards, involving transplants to understand tumor initiation and response to viral treatments, with close monitoring of animal welfare.
Immune characterization of mouse GBMs
Post-treatment, brains were carefully dissected for immune profiling, employing various techniques to dissect cellular compositions and functions.
SABER–FISH detection of transgene copy number
SABER–FISH methods were employed for enhanced imaging and measurement of transgene copies within tissue samples, facilitating detailed examinations.
Assessment of the fraction of the tumour cell population that activates AAV1-SSE-7–mCherry
Actively quantifying cells that respond to viral treatments allowed for the assessment of transgene activation within tumor populations, enhancing understanding of therapeutic impacts.







