








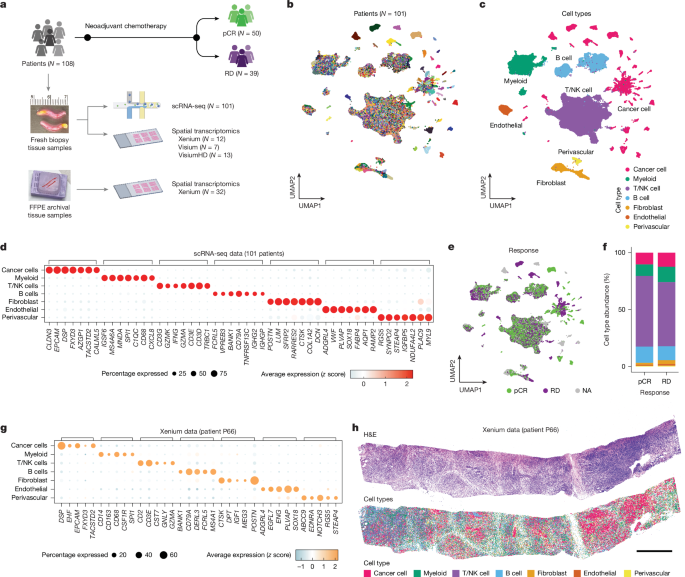
Study participant details from the ARTEMIS Trial
The ARTEMIS Trial’s clinical protocol received approval from The University of Texas MD Anderson Cancer Center’s Institutional Review Board, with all participants giving written informed consent. This was conducted following ethical standards outlined in the 1964 Helsinki declaration and its later amendments. In this study, triple-negative breast cancer (TNBC) was defined as breast cancer that is either estrogen receptor-negative (less than 1%) or low estrogen receptor-positive (1% to less than 10%), as well as progesterone receptor-negative (less than 1%) or low progesterone receptor-positive (1% to less than 10%) and HER2-negative by immunohistochemistry and fluorescence in situ hybridization. The status of residual cancer burden (RCB) after chemotherapy was evaluated through histopathology on the surgically removed specimens. Chemotherapy responses were classified as pathologic complete response (pCR, RCB-0) or residual disease (RD, RCB-I, II or III). A small group of patients (N = 19) who faced a suboptimal mid-treatment response switched to targeted therapies prior to surgery and thus lacked data on neoadjuvant chemotherapy (NAC) response. The gene-expression data from these patients were included for biological analysis, although they were excluded from comparisons of response groups. For each patient, 1–4 fresh tumor breast core needle biopsies were gathered, sent to the lab for cell dissociation, and subsequently frozen for spatial transcriptomic studies.
Experimental method details
scRNA-seq
The biopsies were sectioned into approximately 1 mm³ pieces and digested in a specific dissociation buffer. The resulting cell suspensions underwent a series of processes including centrifugation and trypsin treatment to achieve a cellular pellet, which was then ultimately prepared for single-cell RNA sequencing (scRNA-seq). This involved barcoding and library preparation using established protocols. Details can be found on Protocols.IO.
Visium spatial transcriptomics experiments
For spatial transcriptomics, fresh tumor biopsies were embedded in optimal cutting temperature (OCT) compound, stored at very low temperatures, and sections were prepared for capture on a Visium platform slide. The optimal conditions for breast tissue permeabilization and imaging were determined and adhered to. The final libraries were prepared and sequenced using a next-generation sequencing system.
Xenium in situ RNA experiments
Freshly frozen breast core biopsies were similarly processed and placed on Xenium slides. Custom protocols were applied to ensure effective fixation and gene expression analysis. A panel of 100 genes was developed, enhancing the profiling of the tissue specimens.
VisiumHD spatial transcriptomics experiments
The additional experiments using VisiumHD included embedding tissue, performing imaging, and analyzing H&E staining with modifications tailored to breast tissue samples. Data produced underwent further standard analysis procedures for subsequent studies.
Quantification and statistical analysis
Preprocessing of the scRNA-seq, Visium, VisiumHD and Xenium data
The sequencing reads from the various protocols were demultiplexed and aligned to a human genome reference using recommended pipelines. Surface markers and cell normalization protocols were followed to ensure accurate data representation.
Identification of aneuploid cells from scRNA data
A copy number alteration inference tool was used to identify aneuploid cells based on gene expression data. This involved sampling and analyzing clonal substructures. Results were subject to quality control methods to ensure reliability.
Default scRNA-seq preliminary analysis using Seurat
Data normalization and scaling were performed using standard functions, along with dimensionality reduction and clustering techniques to prepare the data for analysis. This laid the groundwork for identifying different cell types amongst the dataset.
CCA-based integration
To reduce potential batch effects, canonical correlation analysis was employed, integrating data from various batches through anchor identification methods.
DEG analysis
Differentially expressed genes (DEGs) were identified using a combination of existing statistical functions, applying robust testing procedures to ensure significance.
Filtering of scRNA-seq data and identifying main cell types
Poor-quality cells were filtered out based on preset criteria, leading to a more refined analysis of cell types and characteristics within the dataset.
Calculating the frequency of CNAs in a patient cohort
The frequency of copy number alterations was captured from each tumor, using statistical methods to identify significant events across the genome.
Creating pseudo-bulk RNA-seq data
Pseudo-bulk RNA-seq data was created by aggregating UMI counts across specified groups, normalizing these values to maintain consistency in future analyses.
Performing NMF
Non-negative matrix factorization techniques were applied to the processed expression data, with specific parameters tweaked to achieve optimal results.
Determining marker genes of a NMF factor
Marker genes were identified by analyzing the contributions of genes to each NMF factor, employing methods for systematic gene selection based on contribution metrics.
Identifying archetypes
The analysis was narrowed to focus on cancer cells specifically, with a deeper dive into expression profiles to identify distinct archetypes among the analyzed samples.
Measuring archetype specificity
Specificity metrics for archetypes were computed, enabling nuanced understandings of cancer cell behavior and characteristics across samples.
Identifying metaprograms of cancer cells
Subsequent analyses concentrated on transcriptional programs expressed within cells, further refining these into metaprograms to compare cellular behaviors effectively.
Testing the association of archetype and response
Contingency analyses provided insights into the relationships between assigned archetypes and treatment responses, leading to statistical assessments of their significance.
CNA inference in Visium data
Spatial transcriptomics methods helped identify patterns associated with cancer cell prevalence, leading to detailed analyses of the data based on copy number alterations.
Identifying cell types and cell states in Xenium data
A sophisticated label transfer method was employed to correlate the cell identities between scRNA-seq and Xenium data, bolstering analytical robustness.
Identifying cancer-cell bins in VisiumHD data
An integrated analysis of VisiumHD brought crucial insights, using previous data for in-depth identification of cancer cells within the imaging framework.
Gene signature levels in spatial transcriptomics
Gene expression metrics were quantified through various procedures, ensuring that gene signatures were effectively represented within the spatial transcriptomics context.
Calculating module scores of a metaprogram in single cancer cells
Module scores for metaprograms were calculated using the expression data, further centering analyses on individual expression characteristics across different samples.
Assigning cell cycle phase for single cells
Cell cycle phases were assigned based on predefined sets of genes, utilizing standard analytical frameworks tailored for scRNA-seq data.
Integrating TME of TNBC and the normal breast tissues
Data integration between TNBC patients and normal breast tissue was achieved using correlation analysis methods, refining the overall understanding of the tumor microenvironment.
Immune and stromal cell states in scRNA-seq data
A comprehensive approach enabled the subclustering of immune and stromal cells, using informative statistical methods to better categorize and identify cell types.
Single-cell UMAP visualization of cells in scRNA-seq data
Visualization techniques were employed to make sense of individual cell data, allowing for a clearer understanding of the cellular landscape in analyzed samples.
Cell frequency of the metaprograms and TME cell states
Cell frequencies were computed for various metaprograms, adjusting calculations to effectively represent the biological context within samples.
Co-occurrence of cancer cells and TME cells
A co-occurrence network analysis shed light on the interactions between tumor microenvironment cell states and cancer metaprograms, revealing interconnected patterns.
Determining ecotypes of cancer metaprograms and TME cell states
Community detection methods were used to define ecotypes based on analyzed cell states, offering further insights into the makeup of cancer cell communities.
Visualizing the co-occurrence network
Graphical representations were crafted to emphasize the relationships found in the co-occurrence network, utilizing heuristics to create meaningful visual distinctions.
Comparing the co-occurrence network across patient categories
Analyses were conducted to compare networks across different patient categories, providing detailed insights into the shared characteristics and frequencies of cell states.
Ligand–receptor analysis
Using a comprehensive analysis tool, the interactions among ligand-receptor pairs were assessed, contributing to a deeper understanding of cell communications.
Spatial niche analysis in Xenium data
A detailed study on cellular compositions surrounding individual cancer cells was performed, leading to a deeper understanding of spatial interactions within tissue samples.
Selecting patients and treatment arms in public cohort datasets
Validation for the study was achieved by utilizing data derived from various public clinical trials, ensuring a robust comparative analysis between treatment arms.
Predicting the archetypes in external TNBC cohort data
External cohorts were analyzed using internal archetypal data, allowing for a matched prediction process that enhanced understanding of heterogeneity in TNBC.
Survival analysis using the Cox proportional-hazards model
The association of various demographic and clinical variables with overall patient survival was explored, using sophisticated statistical models to derive meaningful conclusions.
Building the cell-state-based classifier to predict NAC response
A predictive model was created focusing on TME cell states and cancer cell programs, employing a mixture of analytical techniques to refine prediction accuracy.
Building the gene-based classifier to predict NAC response
Candidate genes associated with treatment responses underwent rigorous univariate regression analyses, highlighting genes that significantly predicted chemotherapy outcomes.
The gene-based classifier and overall survival
Utilizing established cohorts, survival analyses elucidated relationships between gene expressions and patient outcomes, ensuring a robust understanding of treatment impacts.
Gene signatures of the ‘Vanderbilt’ expression subtypes
Gene expression results were meticulously extracted and processed to develop relevant gene signatures, adding depth to the classification of TNBC subtypes.
Reporting summary
Additional details on research methodology can be found in the linked reporting summary document.







