








Rationale for EBV Detection
The 171,823-nucleotide EBV genome first appeared in December 2013 as part of the hg38 version, serving essentially as a buffer for off-target reads commonly found in sequencing libraries. This was especially relevant for capturing EBV reads linked to the immortalization of LCLs, like those in the 1000 Genomes Project. Notably, whole genome sequencing (WGS) conducted by the UKB and AOU consortia utilized whole blood samples, meaning that any EBV reads identified were likely remnants from earlier infections.
WGS Data and Cohort Analyses in the UKB
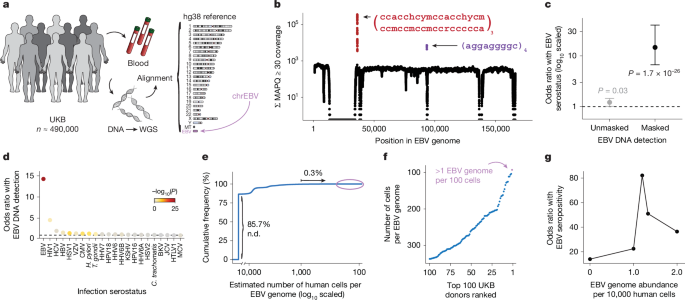
For the UKB, we measured the amount of EBV DNA in 490,560 WGS libraries. This was done by extracting reads that aligned to chrEBV in the hg38 human genome reference, specifically focusing on those with a mapping quality (MAPQ) of 30 or higher. To determine the abundance of EBV DNA at each position, we aggregated the coverage of every base in the EBV genome throughout all libraries. The resulting coverage was mostly even, indicating genuine EBV DNA detection from the WGS reads, but there were notable exceptions. Specifically, 27,692 positions showed little to no coverage due to low mappability, while two regions demonstrated significantly higher coverage levels, suggesting potential confounds in estimating EBV DNA. We assessed these anomalies by measuring EBV DNA abundance before and after masking, focusing on individual donor data. Initial findings showed a weak, though not significant, positive link between EBV DNA presence and seropositivity. However, after accounting for the repetitive regions, the association strengthened considerably.
Contig Mappability Analyses
To investigate why some regions of the EBV contig were not detected, synthetic reads of length 101 bases were generated and aligned. Mappability was defined as the percentage of reads overlapping a position with a MAPQ score over ten. This analysis supported the idea that the lack of detection in certain regions resulted from the hg38 reference’s homology, not from varying DNA presence from previous infections.
EBV DNA Copy Number Estimation
To evaluate EBV DNA per individual, we focused on well-covered, unbiased bases and normalized against the effective EBV genome size, leading to an estimated copy number of 1 in 1,000 to 10,000 cells among people with detectable EBV DNA. When compared to healthy individuals, this was quite lower than the upper limits of EBV copy numbers typically found. In fact, after our calculations, a significant portion (85.7%) of individuals in the UKB showed no detectable EBV DNA, despite over 90% being seropositive.
This prompted a simulation study to delve deeper into the seeming discrepancy. Using maximum likelihood estimation and adjusting certain parameters, we showcased how a single underlying component could explain many observed data features.
Phenome-wide Association Studies
We employed PheWAS using the UKB cohort to assess the connection between EBV DNAemia and numerous phenotypes, both binary and quantitative. Logistic regression was utilized with appropriate corrections for confounding variables like age and sex. To validate findings, we also analyzed the AOU cohort, with a focus on immunosuppressive drug exposure. Interestingly, a positive association was observed, although it was not statistically significant.
Genetic Associations with EBV DNAemia in the UKB
We explored genetic variants linked to EBV DNAemia, carefully stratifying the cohort by various ancestral backgrounds. This yielded important insights into the genetic architecture connecting EBV DNAemia with particular health conditions.
Replication of EBV DNAemia-associated Genotypes
To broaden the understanding of variants responsible for EBV DNAemia, we also utilized data from the AOU, filtering for common variants within European ancestry. Associations were rigorously tested, solidifying the link between certain genetic factors and EBV presence.
Pathway and Single-Cell Analyses
Further studies were undertaken to evaluate gene expression patterns associated with our findings using high-resolution datasets from distinct donors. These analyses helped contextualize the relationships between observed genetic variants and immune responses, aligning perfectly with our previous findings.
EBV Viral Sequence Analysis
Next, we examined raw sequencing reads from EBV across participants, focusing on specific strains and quantifying variance at certain genomic locations. By aligning sequences, we identified several mutations of interest and their potential implications in immune evasion or functional changes.
Conclusion
This elaborative study highlights the importance of precise analyses in estimating EBV DNA presence and recognizes the broader implications for understanding the viral dynamics in human health.







