








Sample acquisition and ethics approval
The University of Pennsylvania’s institutional review board (IRB) authorized the collection of human kidney tissue for this study. The kidney samples collected were completely de-identified, meaning no personal information was recorded, making them exempt from further IRB assessment. An external honest broker managed the clinical data collection while maintaining participant anonymity. None of the participants received compensation. Some kidney samples were sourced from the Transformative Research in Diabetic Nephropathy (TRIDENT) project, which tracks and characterizes individuals with diabetic kidney disease (DKD). This initiative gained approval from both the University of Pennsylvania IRB and the TRIDENT Steering Committee, with informed consent secured from each participant. Additionally, research utilizing the UK Biobank Resource was sanctioned under Application Number 273810, where participants had provided written consent for the use of their samples for medical research.
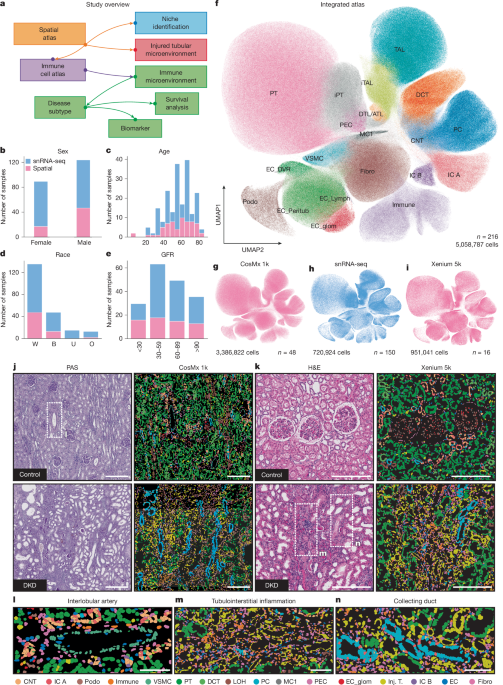
CosMx sample preparation and data preprocessing
Tissue sections of 5 µm thickness were prepared in accordance with the guidelines from NanoString Technologies. Human universal cell characterization RNA probes were employed, along with 50 custom probes targeting various genes. These genes included ESRRB, SLC12A1, UMOD, and others. Staining was conducted using DAPI and specific markers like CD298/B2M, CK8/18, and PanCK/CD45 per established protocols. Imaging was completed with a setup designated as configuration A. Following imaging, slides were treated with 100% xylene and then stained using haematoxylin and eosin. Data from each CosMx run were exported from the AtoMx platform and converted into a Python object for processing. Samples were then preprocessed and analyzed collectively using scanpy, filtering out cells with fewer than 30 counts.
Xenium sample preparation and data preprocessing
Similarly, 5 µm thick tissue sections were prepared on Xenium slides per 10X Genomics’ specifications. The human Xenium Prime 5K Human Pan Tissue and Pathways Panel was utilized, supplemented with 100 additional probes for specific genes, including TPM1, ESRRB, and others. These sections underwent a morphological segmentation workflow followed by transcriptomic analysis via the Xenium Analyzer. Raw output from Xenium was processed using the spatialdata framework, with transcript and segmentation data structured into a SpatialData object. Genes were then filtered to exclude those with fewer than 30 counts.
Integration of the single-cell spatial kidney atlas
To combine single-nucleus RNA sequencing (snRNA-seq) data with spatial transcriptomic data, a deep generative modeling framework was applied using scVI and scANVI, part of the scvi-tools Python package. Two integrations were carried out—one for CosMx data and another for Xenium. The integration utilized raw count matrices and recorded metadata such as technology and sample identifiers. The SCVI.setup_anndata() function was employed for these annotations. The model was trained with a negative binomial likelihood, featuring 4 hidden layers and early stopping based on validation loss. Distance metrics between cells were calculated to discard those with excessive distance. For creating a unified reference map, filtered CosMx and Xenium objects were concatenated, setting tech as a batch key and running further analysis with a refined scANVI model.
Annotation-based niche generation
To assess cell-type-specific neighborhood compositions at varying spatial scales, the number of neighboring cells of each type was measured within 20, 40, 60, and 80 µm radii. Four different neighborhood matrices were constructed reflecting these distances. To capture new neighbor inclusions, differences between successive shells were calculated. Sparse neighborhoods were filtered to ensure better representation. The outcomes were normalized using StandardScaler and dimensionality reduction was conducted through principal component analysis (PCA). The MiniBatchKMeans method helped in clustering the data to identify distinct niches linked to spatial aspects.
COVET and Hotspot analysis
The COVET methodology was utilized to analyze all cells within the CosMx dataset, focusing on those with over 50 neighbors within a 200 µm radius while considering gene expression patterns alongside spatial location. From this, a covariance matrix was generated to capture contextual co-expression details. Similarly, Hotspot analysis identified gene modules across different modalities through cell similarity graphs to quantify gene co-localization. Various techniques were employed to visualize robustly expressed gene correlations in different conditions.
NicheCompass analysis
NicheCompass was employed to examine cellular niches by processing all cells in the CosMx dataset with specific thresholds for neighbor counts while utilizing a graph neural network approach for analysis. Differential gene program testing across latent clusters identified enriched gene programs, visualized via heat maps highlighting niche-specific signatures.
Injured tubular microenvironment generation
This segment concentrated on constructing injured tubular surroundings by assessing the composition of proximal and thick ascending limb cells across various radii, creating neighborhood matrices that encapsulated related cell types. The matrices underwent normalization and clustering to pinpoint niche subtypes, with differential expression analyses identifying shifts between DKD and control samples.
Immune microenvironment generation
For understanding immune cell distributions, neighborhood profiles were built using existing annotations from the kidney immune cell atlas. The compositions of neighbors across various spatial distances were quantified, culminating in a detailed profile for downstream classification. Visualization involved aggregating cell types into broader categories for enhanced interpretation, with specific immune cell types further examined in relation to their presence in different kidney microenvironments under healthy and DKD conditions.
Human kidney immune cell atlas generation
The integration of immune cell data utilized a complex modeling framework to merge spatial transcriptomic with snRNA-seq data. Each immune cell subset was processed separately before being combined and subjected to modeling for improved annotation accuracy. Final clustering results and expression profiles enabled the identification of various immune cell subtypes relevant to kidney health.
B cell atlas generation
B and plasma cell data, sourced from a recent single-cell B cell atlas, were incorporated into a unified atlas, emphasizing refined modeling for accurate type assignment. B cell annotations were drawn from multiple datasets, leveraging deep generative modeling techniques for improved accuracy in identifying subtypes.
Imaging mass spectrometry
The process began with deparaffinizing FFPE tissue sections following traditional protocols. Antigen retrieval and blocking steps were integrated, followed by the application of metal-conjugated antibodies for protein target detection. Imaging mass cytometry was carried out on a special system to achieve detailed cellular profiling and quantitative analysis of detected proteins.
Differentially expressed genes
For assessing differential expression, counts were normalized and analyzed with specialized statistical tests. Results regarding gene expression variations across different patient groups were compiled and evaluated for significance.
Pathway enrichment analysis using Enrichr
To find pathways tied to genes elevated in specific niches, enrichment analysis was employed, ensuring that only those with significant upregulation were examined further.
Pseudobulk differential expression analysis and spatial gene signature derivation
A pseudobulk differential expression analysis was executed to elucidate specific gene signatures within various niches by aggregating raw counts for statistical evaluation. Significant differences were identified, yielding a refined list of upregulated genes for distinct spatial patterns.
Ligand–receptor interactions
Cell–cell communication signals were analyzed using a statistical approach, filtering for significant ligand–receptor interactions specific to healthy and diseased states, thereby concentrating on those most relevant to kidney function.
Bulk RNA-seq correlation
In this final analysis, data processing followed established protocols to measure quality and resolve gene expression patterns. Correlation metrics between spatial gene signatures and fibrosis scores were computed to assess relationships pertinent to kidney conditions.
Hierarchical clustering of bulk RNA kidney samples
Hierarchical clustering was performed on gene expression data, structured according to previously defined immune microenvironment references to identify potential insights.
TRIDENT plasma proteomics analysis
Leveraging a machine learning approach, a classifier was developed to predict kidney outcomes based on proteomic data, assessing factors to enhance the accuracy of survival outcomes in kidney health perspectives.
Validation in UK Biobank cohort
For further validation, analyses were carried out on the UK Biobank cohort, focusing on participants with diabetes-related conditions, exploring how predictive models performed across multiple time points in relation to kidney health.
Immune complex pull-down, LC–MS acquisition and data analysis
Kidney tissue profiling involved immune complex isolation followed by a comprehensive mass spectrometry process to gain insights into protein interactions, with further statistical evaluations to determine expression patterns.
Statistics and reproducibility
The statistical methodologies adopted are laid out in related methods sections, involving robustness checks and validation across multiple platforms to ensure the integrity of findings.
Reporting summary
Detailed information regarding research design can be accessed in the supplementary materials linked to the study.







